Amyvid 1900 Mbq/Ml
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Amyvid 800 MBq/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml injekčního roztoku obsahuje v den a čas kalibrace (ToC) 800 MBq florbetapirum-(18F).
Aktivita obsažená v jedné injekční lahvičce se pohybuje v rozpětí od 800 MBq do 12000 MBq v den a čas kalibrace.
Fluor-18 se s poločasem přibližně 110 minut rozpadá na stabilní kyslík-18 vyzářením pozitronu s energií 634 keV, a následným vyzářením anihilačních fotonů s energií 511 keV.
Pomocné látky se známým účinkem:
Tento léčivý přípravek obsahuje 79 mg/ml ethanolu a až 37 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Tento léčivý přípravek je určen pouze k diagnostickým účelům.
Přípravek Amyvid je radiofarmakum indikované pro vyšetření pozitronovou emisní tomografií (Positron Emission Tomography, PET) k zobrazení denzity P-amyloidních neuritických plaků v mozku dospělých pacientů s kognitivní poruchou, kteří jsou vyšetřováni z hlediska možného onemocnění Alzheimerovou chorobou (Alzheimer’s disease, AD) anebo jiných příčin kognitivní poruchy. Přípravek Amyvid by měl být používán ve spojení s klinickým vyšetřením.
Negativní PET sken obsahuje velmi malé množství plaků nebo žádné plaky, což je stav, který neodpovídá diagnóze AD. Pro omezení v interpretaci pozitivního skenu viz body 4.4 a 5.1.
4.2 Dávkování a způsob podání
PET sken pomocí florbetapiru-(18F) by měl být vyžádán pouze lékařem se zkušenostmi v léčbě neurodegenerativních poruch.
Snímky získané pomocí přípravku Amyvid by měli interpretovat pouze hodnotitelé vyškoleni v hodnocení PET snímků s florbetapirem-(18F). V případech, kdy nelze přesně zjistit polohu šedé hmoty a rozhraní šedé a bílé hmoty na PET skenu se u pacientů doporučuje současně zaznamenaný snímek zhotovený počítačovou tomografií (CT) nebo magnetickou rezonancí (MR), aby vzniknul kombinovaný snímek PET-CT nebo PET-MR (viz bod 4.4 Interpretace snímku).
Dávkování
Doporučená dávka pro dospělého s tělesnou hmotností 70 kg je 370 MBq florbetapiru-(18F). Objem injekce by neměl být menší než 1 ml a ne větší než 10 ml.
Zvláštní skupiny pacientů
Starší populace
Není třeba upravovat dávku v závislosti na věku.
Pacienti s poruchou funkce ledvin a jater
Je třeba pečlivě posoudit aktivitu, která má být podána, protože u těchto pacientů může dojít k zvýšené expozici záření. Viz část 4.4.
Nebyly provedeny extenzivní studie dávkového rozmezí a úpravy dávek léčivého přípravku u normálních a speciálních populací. Farmakokinetika florbetapiru-(18F) u pacientů s poruchou funkce ledvin nebo jater nebyla stanovena.
Pediatrická populace
Neexistuje žádné relevantní použití přípravku Amyvid u pediatrické populace.
Způsob podání K intravenóznímu podání.
K vícedávkovému užití.
Aktivita florbetapiru-(18F) musí být těsně před podáním změřena aktivimetrem (kalibrátor dávky).
Dávka se podává jako injekce intravenózního bolusu, po kterém následuje propláchnutí injekce roztokem chloridu sodného 9 mg/ml (0,9 %) k zajištění podání celé dávky.
Aplikace přípravku Amyvid prostřednictvím krátkého intravenózního katetru (přibližně 4 cm a méně) snižuje potenciální adsorpci účinné látky do katetru.
Aplikace florbetapiru-(18F) musí být intravenózní, aby nedošlo k ozáření v důsledku proniknutí mimo cévu a aby nedošlo k artefaktům při snímkování
Zhotovení snímků
Doporučuje se zhotovit 10ti minutový PET snímek přibližně 30 až 50 minut po intravenózní injekci přípravku Amyvid. Pacienti by měli být uloženi v poloze na zádech s umístěním hlavy v zorném poli PET skeneru tak, aby byl vycentrován mozek, včetně mozečku. K omezení pohybů hlavy je možno použít pásky nebo jiné flexibilní upnutí hlavy. Rekonstrukce by měla zahrnovat korekci útlumu s výslednou velikostí transaxiálních pixelů mezi 2,0 a 3,0 mm.
4.3 Kontraindikace
Hypersenzitivita na účinnou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Zdůvodnění individuálního přínosu/rizika
U každého pacienta musí být expozice záření odůvodněna pravděpodobným přínosem. Aplikovaná aktivita by měla být v každém případě co možná nejnižší, která ještě umožní získat požadované diagnostické informace.
Pacienti s poruchou funkce ledvin a jater
U těchto pacientů je třeba pečlivě zvážit poměr přínos/riziko, protože může dojít k zvýšené expozici záření. Florbetapir-(18F) je vylučován primárně hepatobiliárním systémem, a u pacientů s poruchoz funkce jater je možnost zvýšené expozici záření. Viz též část 4.2.
Pediatrická populace
Pro informace o použití u této populace viz body 4.2 nebo 5.1 Interpretace snímků získaných pomocí přípravku Amyvid
Snímky získané pomocí přípravku Amyvid by měli interpretovat pouze posuzovatelé vyškolení v hodnocení PET snímků s florbetapirem-(18F). Negativní sken zobrazuje velmi malé množství nebo žádné kortikální P-amyloidní plaky. Pozitivní sken zobrazuje střední až častou denzitu. Byly pozorovány chyby v odhadech denzity mozkových neuritických P-amyloidních plaků včetně falešně negativních výsledků.
Hodnocení snímků by mělo probíhat hlavně v transaxiální orientaci, je však třeba mít přístup také k sagitální a koronální rovině. Doporučuje se, aby hodnocení snímků zahrnovalo všechny transaxiální řezy mozku s použitím černobílé stupnice, při maximální intenzitě stupnice nastavené na maximální intenzitu všech mozkových pixelů.
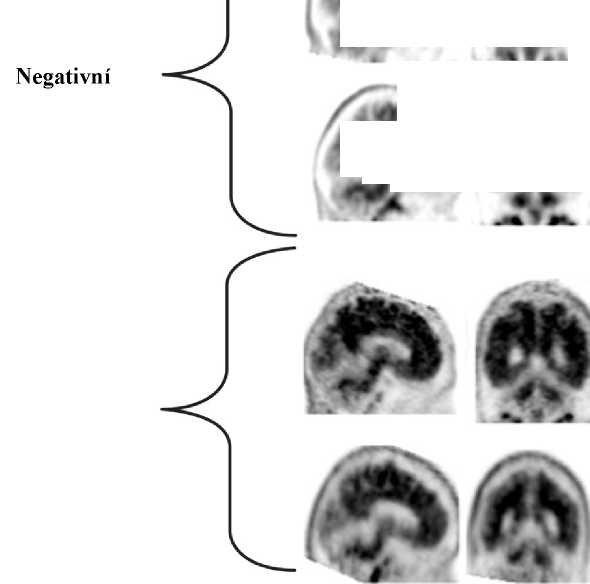
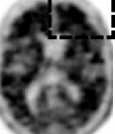
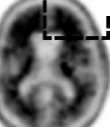
Interpretace negativity nebo pozitivity snímku se provádí vizuálním porovnáním aktivity v šedé hmotě mozkové kůry s aktivitou přilehlé bílé hmoty mozkové kůry (viz obrázek 1).
Negativní skeny mají vyšší aktivitu v bílé hmotě než v šedé hmotě, což vytváří jasný šedo-bílý kontrast. Pozitivní sken bude mít buď:
a) Dvě nebo více mozkových oblastí (každá větší než jeden kortikální gyrus), v nichž je snížený nebo chybějící šedo-bílý kontrast. Toto je nejčastější vzhled pozitivního skenu;
b) Jednu nebo více oblastí, v nichž je aktivita šedé hmoty intenzivní a jasně převyšuje aktivitu v přilehlé bílé hmotě.
Obrázek 1: Případy vyšetření PET při použití přípravku Amyvid ukazují příklady negativních skenů (dvě horní řady) a pozitivních skenů (dolní dvě řady). Panely ve směru zleva doprava ukazují sagitální, koronální a transverzální řezy na PET snímcích. Poslední panel nejvíce vpravo ukazuje zvětšený obraz mozkové oblasti v rámečku. Dvě šipky nahoře ukazují místo normálně zachovaného šedo-bílého kontrastu s kortikální aktivitou, která je nižší než aktivita přilehlé bílé hmoty. Dvě dolní šipky ukazují oblasti sníženého šedo-bílého kontrastu se zvýšenou kortikální aktivitou, která je srovnatelná s aktivitou přilehlé bílé hmoty.
PET snímky získané pomocí přípravku Amyvid
Sagitální
Koronální Transverzální
řh
Pozitivní

\



Omezení při použití
Pozitivní sken není sám osobě dostatečný pro stanovení diagnózy AD nebo jiných kognitivních poruch, protože depozita neuritických plaků v šedé hmotě mohou být přítomna u asymptomatických starších osob a u některých neurodegenerativních demencí (Alzheimerova choroba, demence s Lewyho tělísky, demence při Parkinsonově nemoci).
Pro omezení při použití u pacientů s mírnou kognitivních poruchou (mild cognitive impairment MCI) viz bod 5.1.
Účinnost přípravku Amyvid pro predikci vývoje AD nebo pro monitorování odpovědi na léčbu nebyla prokázána (viz bod 5.1).
Některé skeny mohou být obtížně interpretovatelné v důsledku obrazového šumu, atrofie se ztenčeným pásem kortexu (cortical ribbon) nebo v důsledku rozmazání obrazu, což může vést k chybám při interpretaci. V případech, kdy nelze přesně zjistit umístění šedé hmoty nebo rozhraní šedé a bílé hmoty na PET skenu a je k dispozici současně zaznamenaný snímek zhotovený počítačovou tomografií (CT) nebo magnetickou rezonancí (MR), měl by hodnotitel posoudit kombinovaný snímek PET-CT nebo PET-MR, aby se vyjasnil vztah mezi radioaktivitou v PET a anatomií šedé hmoty.
V některých případech bylo zjištěno zvýšené vychytávání v extracerebrálních strukturách, jako jsou slinné žlázy, kůže, svaly a kosti (viz bod 5.2). Posouzení sagitálních obrazů a současně získaných snímků z CT nebo MR může pomoci rozlišit okcipitální kost od okcipitální šedé hmoty.
Po vyšetření
V průběhu prvních 24 hodin po injekci by měl být omezen blízký kontakt s malými dětmi a těhotnými ženami.
Speciální upozornění
Obsah sodíku je vyšší než 1 mmol (až 37 mg v jedné dávce). To je třeba brát v úvahu u pacientů, kteří drží dietu s nízkým obsahem sodíku.
Tento léčivý přípravek obsahuje 10 objemových % ethanolu (alkoholu), tj. až 790 mg v jedné dávce, což odpovídá 20 ml piva nebo 8 ml vína na dávku.
Toto množství může být škodlivé pro osoby trpící alkoholismem a mělo by být bráno v úvahu u těhotných a kojících žen a u skupin osob s vysokým rizikem jako jsou pacienti s onemocněním jater nebo s epilepsií.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí in vivo.
Vazebné studie in vitro neprokázaly ovlivnění vazby florbetapiru-(18F) k P-amyloidním plakům v přítomnosti dalších obvyklých léčivých přípravků užívaných pacienty s AD.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
V případě plánovaného podání radiofarmak ženě ve fertilním věku je důležité zjistit, zda je nebo není těhotná. U každé ženy, u které se nedostavilo pravidelné menstruační krvácení, je třeba předpokládat, že je těhotná, a to do doby, než se prokáže, že není. V případě pochybností o možném těhotenství u této ženy (pokud se u ní nedostavilo pravidelné menstruační krvácení, pokud jsou menstruační krvácení velmi nepravidelná atd.), by pacientce měly být nabídnuty alternativní techniky nepoužívající ionizující záření (pokud jsou k dispozici).
Postupy a zákroky používající radioaktivní nuklidy prováděné u těhotných žen jsou spojeny s dávkou záření, které je vystaven také plod. V průběhu těhotenství by tedy měla být prováděna pouze nezbytně nutná vyšetření, kdy je pravděpodobný přínos mnohem větší než riziko, kterému jsou vystaveny matka a plod.
Nebyly provedeny žádné studie u těhotných žen. Nebyly provedeny žádné studie u zvířat, které zkoumaly účinky florbetapiru-(18F) na reprodukční funkce (viz bod 5.3).
Kojení
Není známo, zda je florbetapir-(18F) v průběhu kojení vylučován do mateřského mléka. Před podáním radiofarmak matce, která kojí, je třeba uvážit možnost pozdějšího podání radioaktivního nuklidu, tj. vyčkání do doby, kdy matka ukončí kojení, a je třeba také uvážit, jaké nej vhodnější radiofarmakum vzhledem k sekreci aktivity do mateřského mléka. Je-li podání považováno za nezbytné, mělo by být kojení přerušeno na 24 hodin a vytlačené mléko by mělo být zlikvidováno.
V průběhu počátečních 24 hodin po injekci je třeba vyloučit úzký kontakt s malými dětmi.
Fertilita
Nebyly provedeny žádné studie fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje Není relevantní.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Údaje o nežádoucích účincích byly získány v klinických studiích s přípravkem Amyvid, do nichž bylo zařazeno 555 subjektů a v nichž byl přípravek Amyvid jako injekční roztok podán v 665 případech. Nebyly hlášeny žádné závažné nežádoucí účinky související s podáním přípravku Amyvid.
Shrnutí údajů o nežádoucích účincích
Frekvence četnosti nežádoucích účinků jsou definovány následovně: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit). I když se nežádoucí účinky mohou ve skutečnosti vyskytnout v nižších frekvencích, než je uvedeno níže, neumožnila velikost zdrojové databáze provést přiřazení kategorií frekvencí nižších (méně častých resp. vzácnějších) než je kategorie „méně časté“ (>1/1 000 až <1/100).
Poruchy nervového systému Časté: bolest hlavy
Méně časté: disgeuzie (poruchy chuti)
Cévní poruchy
Méně časté: návaly horka
Gastrointestinální poruchy Méně časté: nausea
Poruchy kůže a podkožní tkáně Méně časté: pruritus, kopřivka (urticaria)
Celkové poruchy a reakce v místě aplikace
Méně časté: kožní vyrážka v místě podání infuze, reakce v místě aplikace injekce (zahrnující krvácení v místě injekce, podráždění v místě injekce a bolest v místě injekce)
Expozice ionizačnímu záření je spojena s rozvojem zhoubných nádorů a s rizikem vzniku dědičných vad. Při podání doporučené aktivity 370 MBq florbetapiru-(18F) je odhad účinné dávky přibližně 7 mSv, a proto se očekává, že se tyto nežádoucí účinky objeví s nízkou pravděpodobností.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Vzhledem k malému množství florbetapiru-(18F) v každé dávce se neočekává, že předávkování povede k farmakologickým účinkům. V případě předávkování ozářením by měla být dávka vstřebaná pacientem pokud možno snížena zvýšením eliminace radioaktivního nuklidu z těla častým močením a častou defekací. Může být užitečné odhadnout velikost dávky, která byla aplikována.
FARMAKOLOGICKÉ VLASTNOSTI
5
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: diagnostická radiofarmaka, centrální nervový systém, ATC kód: V09AX05
Mechanismus účinku
Florbetapir-(18F) se váže na neuritické P-amyloidní plaky. Studie vazby látek používající tradiční neuropatologické metody barvení mozků zemřelých pacientů s AD prokázaly statisticky významné (p < 0,0001) korelace mezi vazbou florbetapiru-(18F) in vitro a depozity shluků P -amyloidu.
In vivo byla u pacientů v terminální fázi života posuzována korelace mezi vychytáváním florbetapiru-(18F) v kortikální šedé hmotě a celkovým rozsahem P-amyloidu za použití 4G8 protilátky proti amyloidu, která barví P-amyloid jak v neuritických, tak i difúzních placích. Vazba florbetapiru-(18F) in vivo na jiné P-amyloidní struktury nebo na jiné části mozku nebo receptory zůstává neznámá.
Farmakodynamické účinky
Při nízkých chemických koncentracích přítomných v přípravku Amyvid nemá florbetapir-(18F) žádnou zjistitelnou farmakologickou aktivitu.
V dokončených klinických studiích bylo prováděno kvantitativní měření vychytávání florbetapiru-(18F) v 6 predefinovaných oblastech mozkové kůry (precuneus, frontální, anteriorní cingulum, posteriorní cingulum, parietální a temporální), které bylo měřeno pomocí standardizovaných hodnot vychytávání (standardised uptake values, SUV). Poměry průměrné kortikální SUV (SUVR, ve srovnání s mozečkem) jsou u pacientů s AD vyšší než tyto poměry u zdravých dobrovolníků.
Průměrné hodnoty kortikálního SUVR ve srovnání s mozečkem u pacientů s AD ukazují kontinuální a podstatná zvýšení od času nula až do 30 minut po podání, s pouze malými změnami poté až do 90. minuty od podání injekce. U subjektů léčených obvyklou léčbou pro AD nebyly pozorovány žádné rozdíly ve výsledných hodnotách SUVR ve srovnání se subjekty, kteří žádnou léčbu pro AD nedostávali.
Klinická, účinnost
Pivotní studie u 59 pacientů v terminální fázi života byla zaměřena na stanovení diagnostické účinnosti přípravku Amyvid v detekci denzity kortikálních neuritických plaků (žádná nebo řídká oproti střední nebo časté). Výsledky PET zobrazení byly srovnány s maximální denzitou neuritických plaků v řezech frontální, temporální nebo parietální kůry při autopsii provedené u pacientů do 24 měsíců po PET vyšetření.
Stav kognitivních funkcí pacientů nemohl být spolehlivě měřen. U všech 59 subjektů vedlo zaslepené hodnocení snímků provedené 5 lékaři specialisty v nukleární medicíně ve většině případů k senzitivitě odečtu (read sensitivity) 92% (95% CI: 78-98 %) a specificitě 100 % (95% CI: 80-100 %). Ve studii u 47 mladých (<40 let) zdravých dobrovolníků, u nichž byla předpokládána nepřítomnost P-amyloidu, byly všechny PET snímky s přípravkem Amyvid negativní.
Senzitivita a specificita detekce kortikálního neuritického plaku přípravkem Amyvid byla dále hodnocena ve dvou studiích, v nichž různé skupiny hodnotitelů interpretovali snímky od subjektů sledovaných až do pitvy v pivotní studii. Jejich výsledky úzce korelovaly s výsledky získanými v pivotní studii. Shoda mezi hodnotiteli hodnocená pomocí hodnot Fleissovy kappa byla v rozpětí od 0,75 do 0,85.
V longitudinální studii dále 142 pacientů (klinicky diagnostikovaní jako MCI, AD, nebo s normálními kognitivními funkcemi) podstoupilo základní PET vyšetření florbetapirem-(18F) a bylo sledováno po dobu 3 let, aby byl vyhodnocen vztah mezi zobrazením pomocí přípravku Amyvid a změnami diagnostického stavu.
Diagnostická účinnost vyšetření PET florbetapirem-(18F) PET je uvedena v tabulce níže:
|
Souhlas s počáteční diagnózou MCI N=51 |
Souhlas s počáteční diagnózou klinické AD N=31 | |
|
Senzitivita |
19/51 = 37.3% |
21/31 = 67.7% |
|
(95% CI: 24.1-51.9%) |
(95% CI: 51.3-84.2%) | |
|
Použití non-MCIpřípadů |
Použití non-AD případů | |
|
Specificita |
(kognitivně normální & klinická AD) |
(kognitivně normální & MCI) |
|
69/100 = 69.0% |
91/120 = 75.8% | |
|
(95% CI: 59.9-78.1%) |
(95% CI: 68.2-83.5%) | |
|
Pozitivní pravděpodobnostní poměr (Positive |
1.20 (95% CI: 0.76-1.91) |
2.80 (95% CI: 1.88-4.18) |
|
likelihood ratio) |
U 9 (19%) pacientů, kteří byli klinicky diagnostikováni jako MCI při vstupu do studie, došlo o 36 měsíců později ke konverzi na klinickou AD. 6 (35%) pacientů s MCI ze 17, u kterých byl pozitivní PET sken, byla o 36 měsíců později diagnostikována jako klinicky pravděpodobná AD ve srovnání s 3 (10%) pacienty z 30, u kterých byl sken negativní. Senzitivita skenu s přípravkem Amyvid k prokázání poměru konverze z MCI do AD u 9 konvertovaných pacientů byla 66,7% (95% CI: 35-88%), specificita u 38 nekonvertovaných pacientů byla 71,0% (95% CI: 55-83%) a pozitivní pravděpodobnostní poměr (positive likelihood ratio) byl 2,31 (95% CI: 1.2-4.5). Design této studie nedovoluje stanovení rizika progrese z MCI do klinické AD.
Pediatrická populace
Evropská agentura pro léčivé přípravky upustila od povinnosti předložit výsledky studií použití přípravku Amyvid u všech podskupin v rámci pediatrické populace vzhledem k tomu, že se nepředpokládá použití u pediatrické populace.
5.2 Farmakokinetické vlastnosti
Distribuce v organismu
Florbetapir-(18F) se během několika minut po podání injekce distribuuje do celého organismu a je následně rychle metabolizován.
Vychytávání v orgánech:
K maximálnímu vychytávání florbetapiru-(18F) v mozku dochází během několika minut po podání injekce, s následným rychlým vyplavením z mozku během prvních 30 minut po podání injekce. Orgány s nejvyšší expozicí jsou orgány, v nichž dochází k eliminaci z organismu, zejména žlučník, játra a střeva.
U zdravých kontrol jsou pozorovány poměrně nízké hladiny retence florbetapiru-(18F) v mozkové kůře a v mozečku. Regionální analýzy mozku ukazují mírně vyšší úrovně retence v nucleus caudatum, putamen a v hipokampu. Nejvyšší míra vychytávání je pozorována v oblastech, které jsou tvořeny především bílou hmotou (pons Varoli a centrum semiovale). U subjektů s AD byly pozorovány významně vyšší hodnoty vychytávání v kortikálních oblastech a v putamen než u kontrol. U subjektů s AD dochází, stejně jako u kontrol, k nízké retenci v mozečku a v hipokampu a k vysoké retenci v pons Varoli a v centrum semiovale.
Biofyzikální podklad retence florbetapiru-(18F) v bílé hmotě v živém lidském mozku není možno spolehlivě vysvětlit. Předpokládá se, že k retenci v bílé hmotě může přispívat pomalejší clearance radiofarmaka, protože v bílé hmotě je regionální mozkové prokrvení méně než poloviční oproti mozkové kůře. V některých případech bylo vychytávání pozorováno také v extracerebrálních strukturách, jako je kůže na hlavě, slinné žlázy, svaly a lebeční kost. Důvod tohoto vychytávání není známý, ale může být způsoben akumulací florbetapiru-(18F), některého z jeho radioaktivních metabolitů anebo krevní radioaktivitou.
Eliminace z organismu:
K eliminaci dochází hlavně cestou clearance játry a vylučováním do žlučníku a střev. Je také pozorována určitá akumulace/vylučování cestou močového měchýře. Radioaktivita v moči je přítomna ve formě polárních metabolitů florbetapiru-(18F).
Poločas:
Florbetapir-(18F) je po intravenózní injekci velmi rychle odstraněn z krevního oběhu. Do 20. minuty po podání zůstává v krvi méně než 5 % radioaktivity injikované ve formě fluoru-18, a do 45. minuty po podání jsou přítomna méně než 2 %. Reziduální fluor-18 v krevním oběhu v průběhu 30-90minutového zobrazovacího období je přítomen hlavně ve formě polárních sloučenin fluoru-18. Radioaktivní poločas fluoru-18 je 110 minut.
Pacienti s poruchou funkce ledvin/jater
Farmakokinetika u pacientů s poruchou funkce ledvin nebo jater nebyla stanovena.
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxikologie u zvířat a bezpečnostní farmakologie
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po jednorázovém i opakovaném podání, v nichž byl použit florbetapir [neradioaktivní forma florbetapiru-(18F)], nezjistily žádná speciální rizika pro lidi. Studie s akutními dávkami byla provedena u potkanů, a bylo stanoveno, že NOAEL (no observable adverse effect level, žádná pozorovatelná úroveň nežádoucích účinků) je nejméně 100krát vyšší než maximální dávka u lidí. Potenciální toxicita spojená s podáváním denních opakovaných intravenózních injekcí florbetapiru po dobu 28 dnů byla testována u potkanů a psů, a bylo zjištěno, že NOAEL je nejméně 25krát vyšší než maximální dávka u lidí.
Při testu reverzních bakteriálních mutací in vitro (Ames test) bylo pozorováno zvýšení počtu revertujících kolonií u 2 z 5 kmenů exponovaných florbetapiru. Ve studii chromozomálních aberací in vitro s kultivovanými lidskými periferními lymfocyty nevedl florbetapir ke zvýšení procentního podílu buněk se strukturálními aberacemi při 3hodinové expozici s aktivací nebo bez aktivace; expozice trvající 22 hodin však vyvolala zvýšení strukturálních aberací při všech testovaných koncentracích. Potenciální genotoxicita florbetapiru in vivo byla hodnocena ve studii mikrojader (micronucleus study) u potkanů. V tomto testu nevedl florbetapir při podávání dvakrát denně po 3 po sobě jdoucí dny při nejvyšší dosažitelné dávce 372 ^g/kg/den ke zvýšení počtu polychromatických erytrocytů s mikrojádry. Tato dávka je přibližně 500krát vyšší než maximální dávka u lidí, a nebyly při ní zjištěny žádné známky mutagenicity.
Nebyly provedeny žádné studie s florbetapirem-(18F) u zvířat hodnotící potenciální dlouhodobou karcinogenicitu, fertilitu nebo účinky florbetapiru-(18F) na reprodukci.
S florbetapirem-(18F) nebyly provedeny žádné toxikologické studie a studie bezpečnostní farmakologie.
6 FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Bezvodý ethanol Natrium-askorbát Chlorid sodný Voda pro injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
7,5 hodiny od času provedení kalibrace
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Podmínky uchovávání po prvním otevření viz bod 6.3.
Uchovávejte radiofarmaka v souladu národními právními úpravami pro radioaktivní materiály.
6.5 Druh obalu a obsah balení
Přípravek Amyvid je dodáván v lahvičkách z čirého borosilikátového skla (typ I) o objemu 10 ml nebo 15 ml s chlorobutylovými elastomerovými zátkami pokrytými FluoroTec a hliníkovými uzávěry.
Jedna lahvička pro více dávek s kapacitou 10 ml obsahuje 1 až 10 ml roztoku, což odpovídá 800 až 8000 MBq v den a čas kalibrace.
Jedna lahvička pro více dávek s kapacitou 15 ml obsahuje 1 až 15 ml roztoku, což odpovídá 800 až 12000 MBq v den a čas kalibrace.
V důsledku rozdílů ve výrobním procesu je možné, že lahvičky některých šarží jsou distribuovány s propíchnutými pryžovými zátkami.
Každá lahvička je umístěna ve stíněné nádobě odpovídající tloušťky k omezení expozice zevnímu záření na minimum.
Velikost balení: 1 lahvička.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obecné upozornění:
Radiofarmaka by měly v určeném klinickém prostředí přijímat, používat a podávat pouze oprávněné pověřené osoby za určených klinických podmínek. Příjem, uskladnění, použití, přeprava a likvidace podléhají právním úpravám a/nebo odpovídajícím schválením (licencím) vydaným kompetentním státním úřadem.
Radiofarmaka by měla být připravována způsobem, který je v souladu s požadavky radiační bezpečnosti a farmaceutické kvality. Je třeba učinit odpovídající aseptická bezpečnostní opatření.
Lahvička nesmí být použita, pokud je narušena její celistvost.
Postupy při podávání by měly být prováděny tak, aby bylo riziko kontaminace léčivého přípravku a riziko ozáření obsluhujících osob omezeno na minimum. Je povinné použití odpovídajícího stínění/ochrany.
Podávání radiofarmak je spojeno s rizikem pro další osoby (včetně těhotných zdravotnic) ze zevního ozáření nebo z kontaminace v důsledku rozlití moče, zvratků atd. Je proto nezbytné učinit bezpečnostní opatření pro ochranu před zářením v souladu s národními právními předpisy.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7 DRŽITEL ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht,Nizozemsko
8 REGISTRAČNÍ ČÍSLO(A)
EU/1/12/805/001
EU/1/12/805/002
9 DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
14. ledna 2013
10 DATUM REVIZE TEXTU
11 DOZIMETRIE
Odhad absorbovaných dávek ionizačního záření v orgánech a tkáních u průměrného dospělého pacienta (70 kg) pro 370 MBq florbetapiru-(18F) při použití standardních metod dozimetrických výpočtů (ICRP Ročník 30) je uveden níže v tabulce. Nebyly učiněny žádné předpoklady ohledně vyprázdnění močového měchýře.
|
Orgán/tkáň |
Absorbovaná dávka na jednotku podané aktivity (pGy/MBq) |
|
Průměr | |
|
Nadledviny |
13,6 |
|
Mozek |
10,0 |
|
Prsy |
6,2 |
|
Stěna žlučníku |
143,0 |
|
Stěna dolní části tlustého střeva |
27,8 |
|
Tenké střevo |
65,5 |
|
Stěna žaludku |
11,7 |
|
Stěna horní části tlustého střeva |
74,4 |
|
Srdeční stěna |
12,7 |
|
Ledviny |
13,0 |
|
Játra |
64,4 |
|
Plíce |
8,5 |
|
Svaly |
8,6 |
|
Vaječníky |
17,6 |
|
Slinivka |
14,4 |
|
Červená kostní dřeň |
14,3 |
|
Kostní buňky |
27,6 |
|
Kůže |
5,9 |
|
Slezina |
8,9 |
|
Varlata |
6,8 |
|
Thymus |
7,3 |
|
Štítná žláza |
6,8 |
|
Orgán/tkáň |
Absorbovaná dávka na jednotku podané aktivity (pGy/MBq) |
|
Průměr | |
|
Stěna močového měchýře |
27,1 |
|
Děloha |
15,6 |
|
Celé tělo |
11,6 |
|
Účinná dávka f^Sv/MBq] |
18,6 |
Předpokládán faktor kvality (Q) 1 pro konverzi absorbované dávky na účinnou dávku pro fluor-18.
Účinná dávka po podání dávky 370 MBq pro dospělou osobu s tělesnou hmotností 70 kg je přibližně 7 mSv. Pokud je simultánně prováděno CT snímkování jako součást PET procedury, zvýší se expozice ionizujícímu záření o množství závislé na nastavení použité při získání CT. Pro podanou aktivitu 370 MBq je typická dávka záření do cílového orgánu (mozku) 3,7 mGy.
Pro podanou aktivitu 370 MBq jsou typické dávky záření do kritických orgánů 53 mGy pro žlučník, 27,5 mGy pro stěnu horní části tlustého střeva, 10,3 mGy pro stěnu dolní části tlustého střeva,
24,2 mGy pro tenké střevo a 23,8 mGy pro játra.
12 NÁVOD PRO PŘÍPRAVU RADIOFARMAK
Metoda přípravy
Před použitím musí být balení zkontrolováno a aktivita musí být odpovídajícím způsobem změřena aktivimetrem.
Odběry by měly být provedeny za aseptických podmínek. Lahvičky nesmí být otevřeny před dezinfekcí zátky, roztok by měl být odebrán přes zátku pomocí injekční stříkačky na jedno použití vybavené vhodným ochranným stíněním a sterilní jehlou na jedno použití, nebo pomocí schváleného automatizovaného aplikačního systému. Používejte pouze polypropylenové/HDPE injekční stříkačky. Pokud je narušena celistvost lahvičky, přípravek by neměl být použit.
Přípravek Amyvid může být asepticky naředěn roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce v maximálním ředícím poměru 1:5. Naředěný roztok musí být použit do 4 hodin po naředění,
Kontrola kvality
Před použitím musí být roztok vizuálně zkontrolován. Muže být použit pouze bezbarvý roztok bez viditelných částic.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu/.
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Amyvid 1900 MBq/ml injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden ml injekčního roztoku obsahuje v den a čas kalibrace (ToC) 1900 MBq florbetapirum-(18F).
Aktivita obsažená v jedné injekční lahvičce se pohybuje v rozpětí od 1900 MBq do 28500 MBq v den a čas kalibrace.
Fluor-18 se s poločasem přibližně 110 minut rozpadá na stabilní kyslík-18 vyzářením pozitronu s energií 634 keV, a následným vyzářením anihilačních fotonů s energií 511 keV.
Pomocné látky se známým účinkem:
Tento léčivý přípravek obsahuje 79 mg/ml ethanolu a až 37 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Injekční roztok.
Čirý bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Tento léčivý přípravek je určen pouze k diagnostickým účelům.
Přípravek Amyvid je radiofarmakum indikované pro vyšetření pozitronovou emisní tomografií (Positron Emission Tomography, PET) k zobrazení denzity P-amyloidních neuritických plaků v mozku dospělých pacientů s kognitivní poruchou, kteří jsou vyšetřováni z hlediska možného onemocnění Alzheimerovou chorobou (Alzheimer’s disease, AD) anebo jiných příčin kognitivní poruchy. Přípravek Amyvid by měl být používán ve spojení s klinickým vyšetřením.
Negativní PET sken obsahuje velmi malé množství plaků nebo žádné plaky, což je stav, který neodpovídá diagnóze AD. Pro omezení v interpretaci pozitivního skenu viz body 4.4 a 5.1.
4.2 Dávkování a způsob podání
PET sken pomocí florbetapiru-(18F) by měl být vyžádán pouze lékařem se zkušenostmi v léčbě neurodegenerativních poruch.
Snímky získané pomocí přípravku Amyvid by měli interpretovat pouze hodnotitelé vyškoleni v hodnocení PET snímků s florbetapirem-(18F). V případech, kdy nelze přesně zjistit polohu šedé hmoty a rozhraní šedé a bílé hmoty na PET skenu se u pacientů doporučuje současně zaznamenaný snímek zhotovený počítačovou tomografií (CT) nebo magnetickou rezonancí (MR), aby vzniknul kombinovaný snímek PET-CT nebo PET-MR (viz bod 4.4 Interpretace snímku).
Dávkování
Doporučená dávka pro dospělého s tělesnou hmotností 70 kg je 370 MBq florbetapiru-(18F). Objem injekce by neměl být menší než 1 ml a ne větší než 10 ml.
Zvláštní skupiny pacientů
Starší populace
Není třeba upravovat dávku v závislosti na věku.
Pacienti s poruchou funkce ledvin a jater
Je třeba pečlivě posoudit aktivitu, která má být podána, protože u těchto pacientů může dojít k zvýšené expozici záření. Viz část 4.4.
Nebyly provedeny extenzivní studie dávkového rozmezí a úpravy dávek léčivého přípravku u normálních a speciálních populací. Farmakokinetika florbetapiru-(18F) u pacientů s poruchou funkce ledvin nebo jater nebyla stanovena.
Pediatrická populace
Neexistuje žádné relevantní použití přípravku Amyvid u pediatrické populace.
Způsob podání K intravenóznímu podání.
K vícedávkovému užití.
Aktivita florbetapiru-(18F) musí být těsně před podáním změřena aktivimetrem (kalibrátor dávky).
Dávka se podává jako injekce intravenózního bolusu, po kterém následuje propláchnutí injekce roztokem chloridu sodného 9 mg/ml (0,9 %) k zajištění podání celé dávky.
Aplikace přípravku Amyvid prostřednictvím krátkého intravenózního katetru (přibližně 4 cm a méně) snižuje potenciální adsorpci účinné látky do katetru.
Aplikace florbetapiru-(18F) musí být intravenózní, aby nedošlo k ozáření v důsledku proniknutí mimo cévu a aby nedošlo k artefaktům při snímkování
Zhotovení snímků
Doporučuje se zhotovit 10ti minutový PET snímek přibližně 30 až 50 minut po intravenózní injekci přípravku Amyvid. Pacienti by měli být uloženi v poloze na zádech s umístěním hlavy v zorném poli PET skeneru tak, aby byl vycentrován mozek, včetně mozečku. K omezení pohybů hlavy je možno použít pásky nebo jiné flexibilní upnutí hlavy. Rekonstrukce by měla zahrnovat korekci útlumu s výslednou velikostí transaxiálních pixelů mezi 2,0 a 3,0 mm.
4.3 Kontraindikace
Hypersenzitivita na účinnou látku nebo na kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Zdůvodnění individuálního přínosu/rizika
U každého pacienta musí být expozice záření odůvodněna pravděpodobným přínosem. Aplikovaná aktivita by měla být v každém případě co možná nejnižší, která ještě umožní získat požadované diagnostické informace.
Pacienti s poruchou funkce ledvin a jater
U těchto pacientů je třeba pečlivě zvážit poměr přínos/riziko, protože může dojít k zvýšené expozici záření. Florbetapir-(18F) je vylučován primárně hepatobiliárním systémem, a u pacientů s poruchoz funkce jater je možnost zvýšené expozici záření. Viz též část 4.2.
Pediatrická populace
Pro informace o použití u této populace viz body 4.2 nebo 5.1 Interpretace snímků získaných pomocí přípravku Amyvid
Snímky získané pomocí přípravku Amyvid by měli interpretovat pouze posuzovatelé vyškolení v hodnocení PET snímků s florbetapirem-(18F). Negativní sken zobrazuje velmi malé množství nebo žádné kortikální P-amyloidní plaky. Pozitivní sken zobrazuje střední až častou denzitu. Byly pozorovány chyby v odhadech denzity mozkových neuritických P-amyloidních plaků včetně falešně negativních výsledků.
Hodnocení snímků by mělo probíhat hlavně v transaxiální orientaci, je však třeba mít přístup také k sagitální a koronální rovině. Doporučuje se, aby hodnocení snímků zahrnovalo všechny transaxiální řezy mozku s použitím černobílé stupnice, při maximální intenzitě stupnice nastavené na maximální intenzitu všech mozkových pixelů.
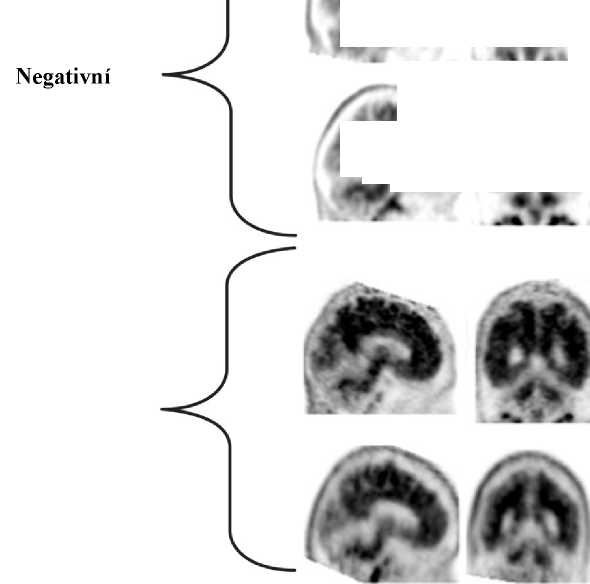
Interpretace negativity nebo pozitivity snímku se provádí vizuálním porovnáním aktivity v šedé hmotě mozkové kůry s aktivitou přilehlé bílé hmoty mozkové kůry (viz obrázek 1).
Negativní skeny mají vyšší aktivitu v bílé hmotě než v šedé hmotě, což vytváří jasný šedo-bílý kontrast. Pozitivní sken bude mít buď:
a) Dvě nebo více mozkových oblastí (každá větší než jeden kortikální gyrus), v nichž je snížený nebo chybějící šedo-bílý kontrast. Toto je nejčastější vzhled pozitivního skenu; b) Jednu nebo více oblastí, v nichž je aktivita šedé hmoty intenzivní a jasně převyšuje aktivitu v přilehlé bílé hmotě.
Obrázek 1: Případy vyšetření PET při použití přípravku Amyvid ukazují příklady negativních skenů (dvě horní řady) a pozitivních skenů (dolní dvě řady). Panely ve směru zleva doprava ukazují sagitální, koronální a transverzální řezy na PET snímcích. Poslední panel nejvíce vpravo ukazuje zvětšený obraz mozkové oblasti v rámečku. Dvě šipky nahoře ukazují místo normálně zachovaného šedo-bílého kontrastu s kortikální aktivitou, která je nižší než aktivita přilehlé bílé hmoty. Dvě dolní šipky ukazují oblasti sníženého šedo-bílého kontrastu se zvýšenou kortikální aktivitou, která je srovnatelná s aktivitou přilehlé bílé hmoty.
PET snímky získané pomocí přípravku Amyvid
Sagitální
Koronální Transverzální
řh
Pozitivní



\
%

Omezení při použití
Pozitivní sken není sám osobě dostatečný pro stanovení diagnózy AD nebo jiných kognitivních poruch, protože depozita neuritických plaků v šedé hmotě mohou být přítomna u asymptomatických starších osob a u některých neurodegenerativních demencí (Alzheimerova choroba, demence s Lewyho tělísky, demence při Parkinsonově nemoci).
Pro omezení při použití u pacientů s mírnou kognitivních poruchou (mild cognitive impairment MCI) viz bod 5.1.
Účinnost přípravku Amyvid pro predikci vývoje AD nebo pro monitorování odpovědi na léčbu nebyla prokázána (viz bod 5.1).
Některé skeny mohou být obtížně interpretovatelné v důsledku obrazového šumu, atrofie se ztenčeným pásem kortexu (cortical ribbon) nebo v důsledku rozmazání obrazu, což může vést k chybám při interpretaci. V případech, kdy nelze přesně zjistit umístění šedé hmoty nebo rozhraní šedé a bílé hmoty na PET skenu a je k dispozici současně zaznamenaný snímek zhotovený počítačovou tomografií (CT) nebo magnetickou rezonancí (MR), měl by hodnotitel posoudit kombinovaný snímek PET-CT nebo PET-MR, aby se vyjasnil vztah mezi radioaktivitou v PET a anatomií šedé hmoty.
V některých případech bylo zjištěno zvýšené vychytávání v extracerebrálních strukturách, jako jsou slinné žlázy, kůže, svaly a kosti (viz bod 5.2). Posouzení sagitálních obrazů a současně získaných snímků z CT nebo MR může pomoci rozlišit okcipitální kost od okcipitální šedé hmoty.
Po vyšetření
V průběhu prvních 24 hodin po injekci by měl být omezen blízký kontakt s malými dětmi a těhotnými ženami.
Speciální upozornění
Obsah sodíku je vyšší než 1 mmol (až 37 mg v jedné dávce). To je třeba brát v úvahu u pacientů, kteří drží dietu s nízkým obsahem sodíku.
Tento léčivý přípravek obsahuje 10 objemových % ethanolu (alkoholu), tj. až 790 mg v jedné dávce, což odpovídá 20 ml piva nebo 8 ml vína na dávku.
Toto množství může být škodlivé pro osoby trpící alkoholismem a mělo by být bráno v úvahu u těhotných a kojících žen a u skupin osob s vysokým rizikem jako jsou pacienti s onemocněním jater nebo s epilepsií.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly provedeny žádné studie interakcí in vivo.
Vazebné studie in vitro neprokázaly ovlivnění vazby florbetapiru-(18F) k P-amyloidním plakům v přítomnosti dalších obvyklých léčivých přípravků užívaných pacienty s AD.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
V případě plánovaného podání radiofarmak ženě ve fertilním věku je důležité zjistit, zda je nebo není těhotná. U každé ženy, u které se nedostavilo pravidelné menstruační krvácení, je třeba předpokládat, že je těhotná, a to do doby, než se prokáže, že není. V případě pochybností o možném těhotenství u této ženy (pokud se u ní nedostavilo pravidelné menstruační krvácení, pokud jsou menstruační krvácení velmi nepravidelná atd.), by pacientce měly být nabídnuty alternativní techniky nepoužívající ionizující záření (pokud jsou k dispozici).
Postupy a zákroky používající radioaktivní nuklidy prováděné u těhotných žen jsou spojeny s dávkou záření, které je vystaven také plod. V průběhu těhotenství by tedy měla být prováděna pouze nezbytně nutná vyšetření, kdy je pravděpodobný přínos mnohem větší než riziko, kterému jsou vystaveny matka a plod.
Nebyly provedeny žádné studie u těhotných žen. Nebyly provedeny žádné studie u zvířat, které zkoumaly účinky florbetapiru-(18F) na reprodukční funkce (viz bod 5.3).
Kojení
Není známo, zda je florbetapir-(18F) v průběhu kojení vylučován do mateřského mléka. Před podáním radiofarmak matce, která kojí, je třeba uvážit možnost pozděj šího podání radioaktivního nuklidu, tj. vyčkání do doby, kdy matka ukončí kojení, a je třeba také uvážit, jaké nejvhodnější radiofarmakum vzhledem k sekreci aktivity do mateřského mléka. Je-li podání považováno za nezbytné, mělo by být kojení přerušeno na 24 hodin a vytlačené mléko by mělo být zlikvidováno.
V průběhu počátečních 24 hodin po injekci je třeba vyloučit úzký kontakt s malými dětmi.
Fertilita
Nebyly provedeny žádné studie fertility.
4.7 Účinky na schopnost řídit a obsluhovat stroje Není relevantní.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Údaje o nežádoucích účincích byly získány v klinických studiích s přípravkem Amyvid, do nichž bylo zařazeno 555 subjektů a v nichž byl přípravek Amyvid jako injekční roztok podán v 665 případech. Nebyly hlášeny žádné závažné nežádoucí účinky související s podáním přípravku Amyvid.
Shrnutí údajů o nežádoucích účincích
Frekvence četnosti nežádoucích účinků jsou definovány následovně: velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit). I když se nežádoucí účinky mohou ve skutečnosti vyskytnout v nižších frekvencích, než je uvedeno níže, neumožnila velikost zdrojové databáze provést přiřazení kategorií frekvencí nižších (méně častých resp. vzácnějších) než je kategorie „méně časté“ (>1/1 000 až <1/100).
Poruchy nervového systému Časté: bolest hlavy
Méně časté: disgeuzie (poruchy chuti)
Cévní poruchy
Méně časté: návaly horka
Gastrointestinální poruchy Méně časté: nausea
Poruchy kůže a podkožní tkáně Méně časté: pruritus, kopřivka (urticaria)
Celkové poruchy a reakce v místě aplikace
Méně časté: kožní vyrážka v místě podání infuze, reakce v místě aplikace injekce (zahrnující krvácení v místě injekce, podráždění v místě injekce a bolest v místě injekce)
Expozice ionizačnímu záření je spojena s rozvojem zhoubných nádorů a s rizikem vzniku dědičných vad. Při podání doporučené aktivity 370 MBq florbetapiru-(18F) je odhad účinné dávky přibližně 7 mSv, a proto se očekává, že se tyto nežádoucí účinky objeví s nízkou pravděpodobností.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Vzhledem k malému množství florbetapiru-(18F) v každé dávce se neočekává, že předávkování povede k farmakologickým účinkům. V případě předávkování ozářením by měla být dávka vstřebaná pacientem pokud možno snížena zvýšením eliminace radioaktivního nuklidu z těla častým močením a častou defekací. Může být užitečné odhadnout velikost dávky, která byla aplikována.
FARMAKOLOGICKÉ VLASTNOSTI
5
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: diagnostická radiofarmaka, centrální nervový systém, ATC kód: V09AX05
Mechanismus účinku
Florbetapir-(18F) se váže na neuritické P-amyloidní plaky. Studie vazby látek používající tradiční neuropatologické metody barvení mozků zemřelých pacientů s AD prokázaly statisticky významné (p < 0,0001) korelace mezi vazbou florbetapiru-(18F) in vitro a depozity shluků P -amyloidu.
In vivo byla u pacientů v terminální fázi života posuzována korelace mezi vychytáváním florbetapiru-(18F) v kortikální šedé hmotě a celkovým rozsahem P-amyloidu za použití 4G8 protilátky proti amyloidu, která barví P-amyloid jak v neuritických, tak i difúzních placích. Vazba florbetapiru-(18F) in vivo na jiné P-amyloidní struktury nebo na jiné části mozku nebo receptory zůstává neznámá.
Farmakodynamické účinky
Při nízkých chemických koncentracích přítomných v přípravku Amyvid nemá florbetapir-(18F) žádnou zjistitelnou farmakologickou aktivitu.
V dokončených klinických studiích bylo prováděno kvantitativní měření vychytávání florbetapiru-(18F) v 6 predefinovaných oblastech mozkové kůry (precuneus, frontální, anteriorní cingulum, posteriorní cingulum, parietální a temporální), které bylo měřeno pomocí standardizovaných hodnot vychytávání (standardised uptake values, SUV). Poměry průměrné kortikální SUV (SUVR, ve srovnání s mozečkem) jsou u pacientů s AD vyšší než tyto poměry u zdravých dobrovolníků.
Průměrné hodnoty kortikálního SUVR ve srovnání s mozečkem u pacientů s AD ukazují kontinuální a podstatná zvýšení od času nula až do 30 minut po podání, s pouze malými změnami poté až do 90. minuty od podání injekce. U subjektů léčených obvyklou léčbou pro AD nebyly pozorovány žádné rozdíly ve výsledných hodnotách SUVR ve srovnání se subjekty, kteří žádnou léčbu pro AD nedostávali.
Klinická, účinnost
Pivotní studie u 59 pacientů v terminální fázi života byla zaměřena na stanovení diagnostické účinnosti přípravku Amyvid v detekci denzity kortikálních neuritických plaků (žádná nebo řídká oproti střední nebo časté). Výsledky PET zobrazení byly srovnány s maximální denzitou neuritických plaků v řezech frontální, temporální nebo parietální kůry při autopsii provedené u pacientů do 24 měsíců po PET vyšetření.
Stav kognitivních funkcí pacientů nemohl být spolehlivě měřen. U všech 59 subjektů vedlo zaslepené hodnocení snímků provedené 5 lékaři specialisty v nukleární medicíně ve většině případů k senzitivitě odečtu (read sensitivity) 92% (95% CI: 78-98 %) a specificitě 100 % (95% CI: 80-100 %). Ve studii u 47 mladých (<40 let) zdravých dobrovolníků, u nichž byla předpokládána nepřítomnost P-amyloidu, byly všechny PET snímky s přípravkem Amyvid negativní.
Senzitivita a specificita detekce kortikálního neuritického plaku přípravkem Amyvid byla dále hodnocena ve dvou studiích, v nichž různé skupiny hodnotitelů interpretovali snímky od subjektů sledovaných až do pitvy v pivotní studii. Jejich výsledky úzce korelovaly s výsledky získanými v pivotní studii. Shoda mezi hodnotiteli hodnocená pomocí hodnot Fleissovy kappa byla v rozpětí od 0,75 do 0,85.
V longitudinální studii dále 142 pacientů (klinicky diagnostikovaní jako MCI, AD, nebo s normálními kognitivními funkcemi) podstoupilo základní PET vyšetření florbetapirem-(18F) a bylo sledováno po dobu 3 let, aby byl vyhodnocen vztah mezi zobrazením pomocí přípravku Amyvid a změnami diagnostického stavu.
Diagnostická účinnost vyšetření PET florbetapirem-(18F) PET je uvedena v tabulce níže:
|
Souhlas s počáteční diagnózou MCI N=51 |
Souhlas s počáteční diagnózou klinické AD N=31 | |
|
Senzitivita |
19/51 = 37.3% |
21/31 = 67.7% |
|
(95% CI: 24.1-51.9%) |
(95% CI: 51.3-84.2%) | |
|
Použití non-MCIpřípadů |
Použití non-AD případů | |
|
Specificita |
(kognitivně normální & klinická AD) |
(kognitivně normální & MCI) |
|
69/100 = 69.0% |
91/120 = 75.8% | |
|
(95% CI: 59.9-78.1%) |
(95% CI: 68.2-83.5%) | |
|
Pozitivní pravděpodobnostní poměr (Positive |
1.20 (95% CI: 0.76-1.91) |
2.80 (95% CI: 1.88-4.18) |
|
likelihood ratio) |
U 9 (19%) pacientů, kteří byli klinicky diagnostikováni jako MCI při vstupu do studie, došlo o 36 měsíců později ke konverzi na klinickou AD. 6 (35%) pacientů s MCI ze 17, u kterých byl pozitivní PET sken, byla o 36 měsíců později diagnostikována jako klinicky pravděpodobná AD ve srovnání s 3 (10%) pacienty z 30, u kterých byl sken negativní. Senzitivita skenu s přípravkem Amyvid k prokázání poměru konverze z MCI do AD u 9 konvertovaných pacientů byla 66,7% (95% CI: 35-88%), specificita u 38 nekonvertovaných pacientů byla 71,0% (95% CI: 55-83%) a pozitivní pravděpodobnostní poměr (positive likelihood ratio) byl 2,31 (95% CI: 1.2-4.5). Design této studie nedovoluje stanovení rizika progrese z MCI do klinické AD.
Pediatrická populace
Evropská agentura pro léčivé přípravky upustila od povinnosti předložit výsledky studií použití přípravku Amyvid u všech podskupin v rámci pediatrické populace vzhledem k tomu, že se nepředpokládá použití u pediatrické populace.
5.2 Farmakokinetické vlastnosti
Distribuce v organismu
Florbetapir-(18F) se během několika minut po podání injekce distribuuje do celého organismu a je následně rychle metabolizován.
Vychytávání v orgánech:
K maximálnímu vychytávání florbetapiru-(18F) v mozku dochází během několika minut po podání injekce, s následným rychlým vyplavením z mozku během prvních 30 minut po podání injekce. Orgány s nejvyšší expozicí jsou orgány, v nichž dochází k eliminaci z organismu, zejména žlučník, játra a střeva.
U zdravých kontrol jsou pozorovány poměrně nízké hladiny retence florbetapiru-(18F) v mozkové kůře a v mozečku. Regionální analýzy mozku ukazují mírně vyšší úrovně retence v nucleus caudatum, putamen a v hipokampu. Nejvyšší míra vychytávání je pozorována v oblastech, které jsou tvořeny především bílou hmotou (pons Varoli a centrum semiovale). U subjektů s AD byly pozorovány významně vyšší hodnoty vychytávání v kortikálních oblastech a v putamen než u kontrol. U subjektů s AD dochází, stejně jako u kontrol, k nízké retenci v mozečku a v hipokampu a k vysoké retenci v pons Varoli a v centrum semiovale.
Biofyzikální podklad retence florbetapiru-(18F) v bílé hmotě v živém lidském mozku není možno spolehlivě vysvětlit. Předpokládá se, že k retenci v bílé hmotě může přispívat pomalejší clearance radiofarmaka, protože v bílé hmotě je regionální mozkové prokrvení méně než poloviční oproti mozkové kůře. V některých případech bylo vychytávání pozorováno také v extracerebrálních strukturách, jako je kůže na hlavě, slinné žlázy, svaly a lebeční kost. Důvod tohoto vychytávání není známý, ale může být způsoben akumulací florbetapiru-(18F), některého z jeho radioaktivních metabolitů anebo krevní radioaktivitou.
Eliminace z organismu:
K eliminaci dochází hlavně cestou clearance játry a vylučováním do žlučníku a střev. Je také pozorována určitá akumulace/vylučování cestou močového měchýře. Radioaktivita v moči je přítomna ve formě polárních metabolitů florbetapiru-(18F).
Poločas:
Florbetapir-(18F) je po intravenózní injekci velmi rychle odstraněn z krevního oběhu. Do 20. minuty po podání zůstává v krvi méně než 5 % radioaktivity injikované ve formě fluoru-18, a do 45. minuty po podání jsou přítomna méně než 2 %. Reziduální fluor-18 v krevním oběhu v průběhu 30-90minutového zobrazovacího období je přítomen hlavně ve formě polárních sloučenin fluoru-18. Radioaktivní poločas fluoru-18 je 110 minut.
Pacienti s poruchou funkce ledvin/jater
Farmakokinetika u pacientů s poruchou funkce ledvin nebo jater nebyla stanovena.
5.3 Předklinické údaje vztahující se k bezpečnosti
Toxikologie u zvířat a bezpečnostní farmakologie
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti a toxicity po jednorázovém i opakovaném podání, v nichž byl použit florbetapir [neradioaktivní forma florbetapiru-(18F)], nezjistily žádná speciální rizika pro lidi. Studie s akutními dávkami byla provedena u potkanů, a bylo stanoveno, že NOAEL (no observable adverse effect level, žádná pozorovatelná úroveň nežádoucích účinků) je nejméně 100krát vyšší než maximální dávka u lidí. Potenciální toxicita spojená s podáváním denních opakovaných intravenózních injekcí florbetapiru po dobu 28 dnů byla testována u potkanů a psů, a bylo zjištěno, že NOAEL je nejméně 25krát vyšší než maximální dávka u lidí.
Při testu reverzních bakteriálních mutací in vitro (Ames test) bylo pozorováno zvýšení počtu revertujících kolonií u 2 z 5 kmenů exponovaných florbetapiru. Ve studii chromozomálních aberací in vitro s kultivovanými lidskými periferními lymfocyty nevedl florbetapir ke zvýšení procentního podílu buněk se strukturálními aberacemi při 3hodinové expozici s aktivací nebo bez aktivace; expozice trvající 22 hodin však vyvolala zvýšení strukturálních aberací při všech testovaných koncentracích. Potenciální genotoxicita florbetapiru in vivo byla hodnocena ve studii mikrojader (micronucleus study) u potkanů. V tomto testu nevedl florbetapir při podávání dvakrát denně po 3 po sobě jdoucí dny při nejvyšší dosažitelné dávce 372 pg/kg/den ke zvýšení počtu polychromatických erytrocytů s mikrojádry. Tato dávka je přibližně 500krát vyšší než maximální dávka u lidí, a nebyly při ní zjištěny žádné známky mutagenicity.
Nebyly provedeny žádné studie s florbetapirem-(18F) u zvířat hodnotící potenciální dlouhodobou karcinogenicitu, fertilitu nebo účinky florbetapiru-(18F) na reprodukci.
S florbetapirem-(18F) nebyly provedeny žádné toxikologické studie a studie bezpečnostní farmakologie.
6 FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Bezvodý ethanol Natrium-askorbát Chlorid sodný Voda pro injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s žádnými dalšími léčivými přípravky.
6.3 Doba použitelnosti
10 hodin od času provedení kalibrace.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Podmínky uchovávání po prvním otevření viz bod 6.3.
Uchovávejte radiofarmaka v souladu národními právními úpravami pro radioaktivní materiály.
6.5 Druh obalu a obsah balení
Přípravek Amyvid je dodáván v lahvičkách z čirého borosilikátového skla (typ I) o objemu 10 ml nebo 15 ml s chlorobutylovými elastomerovými zátkami pokrytými FluoroTec a hliníkovými uzávěry.
Jedna lahvička pro více dávek s kapacitou 10 ml obsahuje 1 až 10 ml roztoku, což odpovídá 1900 až 19000 MBq v den a čas kalibrace.
Jedna lahvička pro více dávek s kapacitou 15 ml obsahuje 1 až 15 ml roztoku, což odpovídá 1900 až 28500 MBq v den a čas kalibrace.
V důsledku rozdílů ve výrobním procesu je možné, že lahvičky některých šarží jsou distribuovány s propíchnutými pryžovými zátkami.
Každá lahvička je umístěna ve stíněné nádobě odpovídající tloušťky k omezení expozice zevnímu záření na minimum.
Velikost balení: 1 lahvička.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obecné upozornění:
Radiofarmaka by měly v určeném klinickém prostředí přijímat, používat a podávat pouze oprávněné pověřené osoby za určených klinických podmínek. Příjem, uskladnění, použití, přeprava a likvidace podléhají právním úpravám a/nebo odpovídajícím schválením (licencím) vydaným kompetentním státním úřadem.
Radiofarmaka by měla být připravována způsobem, který je v souladu s požadavky radiační bezpečnosti a farmaceutické kvality. Je třeba učinit odpovídající aseptická bezpečnostní opatření.
Lahvička nesmí být použita, pokud je narušena její celistvost.
Postupy při podávání by měly být prováděny tak, aby bylo riziko kontaminace léčivého přípravku a riziko ozáření obsluhujících osob omezeno na minimum. Je povinné použití odpovídajícího stínění/ochrany.
Podávání radiofarmak je spojeno s rizikem pro další osoby (včetně těhotných zdravotnic) ze zevního ozáření nebo z kontaminace v důsledku rozlití moče, zvratků atd. Je proto nezbytné učinit bezpečnostní opatření pro ochranu před zářením v souladu s národními právními předpisy.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7 DRŽITEL ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Nizozemsko
8 REGISTRAČNÍ ČÍSLO(A)
EU/1/12/805/003
EU/1/12/805/004
9 DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
14. ledna 2013
10 DATUM REVIZE TEXTU
11 DOZIMETRIE
Odhad absorbovaných dávek ionizačního záření v orgánech a tkáních u průměrného dospělého pacienta (70 kg) pro 370 MBq florbetapiru-(18F) při použití standardních metod dozimetrických výpočtů (ICRP Ročník 30) je uveden níže v tabulce. Nebyly učiněny žádné předpoklady ohledně vyprázdnění močového měchýře.
|
Orgán/tkáň |
Absorbovaná dávka na jednotku podané aktivity (pGy/MBq) |
|
Průměr | |
|
Nadledviny |
13,6 |
|
Mozek |
10,0 |
|
Prsy |
6,2 |
|
Stěna žlučníku |
143,0 |
|
Stěna dolní části tlustého střeva |
27,8 |
|
Tenké střevo |
65,5 |
|
Stěna žaludku |
11,7 |
|
Stěna horní části tlustého střeva |
74,4 |
|
Srdeční stěna |
12,7 |
|
Ledviny |
13,0 |
|
Játra |
64,4 |
|
Plíce |
8,5 |
|
Svaly |
8,6 |
|
Vaječníky |
17,6 |
|
Slinivka |
14,4 |
|
Červená kostní dřeň |
14,3 |
|
Kostní buňky |
27,6 |
|
Kůže |
5,9 |
|
Slezina |
8,9 |
|
Varlata |
6,8 |
|
Thymus |
7,3 |
|
Štítná žláza |
6,8 |
|
Orgán/tkáň |
Absorbovaná dávka na jednotku podané aktivity (pGy/MBq) |
|
Průměr | |
|
Stěna močového měchýře |
27,1 |
|
Děloha |
15,6 |
|
Celé tělo |
11,6 |
|
Účinná dávka [ySv/MBq] |
18,6 |
Předpokládán faktor kvality (Q) 1 pro konverzi absorbované dávky na účinnou dávku pro fluor-18.
Účinná dávka po podání dávky 370 MBq pro dospělou osobu s tělesnou hmotností 70 kg je přibližně 7 mSv. Pokud je simultánně prováděno CT snímkování jako součást PET procedury, zvýší se expozice ionizujícímu záření o množství závislé na nastavení použité při získání CT. Pro podanou aktivitu 370 MBq je typická dávka záření do cílového orgánu (mozku) 3,7 mGy.
Pro podanou aktivitu 370 MBq jsou typické dávky záření do kritických orgánů 53 mGy pro žlučník, 27,5 mGy pro stěnu horní části tlustého střeva, 10,3 mGy pro stěnu dolní části tlustého střeva,
24,2 mGy pro tenké střevo a 23,8 mGy pro játra.
12 NÁVOD PRO PŘÍPRAVU RADIOFARMAK
Metoda přípravy
Před použitím musí být balení zkontrolováno a aktivita musí být odpovídajícím způsobem změřena aktivimetrem.
Odběry by měly být provedeny za aseptických podmínek. Lahvičky nesmí být otevřeny před dezinfekcí zátky, roztok by měl být odebrán přes zátku pomocí injekční stříkačky na jedno použití vybavené vhodným ochranným stíněním a sterilní jehlou na jedno použití, nebo pomocí schváleného automatizovaného aplikačního systému. Používejte pouze polypropylenové/HDPE injekční stříkačky. Pokud je narušena celistvost lahvičky, přípravek by neměl být použit.
Přípravek Amyvid může být asepticky naředěn roztokem chloridu sodného 9 mg/ml (0,9%) pro injekce v maximálním ředícím poměru 1:5. Naředěný roztok musí být použit do 4 hodin po naředění,
Kontrola kvality
Před použitím musí být roztok vizuálně zkontrolován. Muže být použit pouze bezbarvý roztok bez viditelných částic.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu/.
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Advanced Accelerator Applications-Béthune 126 Rocade Sud 62660 Beuvry Francie
Advanced Accelerator Applications - Saint Genis-Pouilly
20 Rue Diesel
01630 Saint Genis-Pouilly
Francie
Advanced Accelerator Applications (Italy), S.r.l. - Ivrea Via Ribes 5
10010 Colleretto Giacosa (TO)
Itálie
Advanced Accelerator Applications (Italy), S.r.l. - Venafro Via Dell’Industria 86077 Pozzilli (IS)
Itálie
Advanced Accelerator Applications (Italy), S.r.l.
Via Piero Maroncelli 40 47014 Meldola (FC)
Itálie
Advanced Accelerator Applications Ibérica Avda Navarra 3-5 Pol. Ind. La Cuesta, Sector 3 50100 La Almunia de Dona Godina Zaragoza Španělsko
Laboratories Cyclopharma - Glisy 7 Allée Nautilus ZAC Croix de Fer
80440 Glisy Francie
Laboratories Cyclopharma - Toulouse
3 Place Pierre Potier
Oncopole de Toulouse
31000 Toulouse-Langlade
Francie
PETNET Solutions
Heathfield Way, Nottingham City Hospital, Gate 1
Hucknall Road
Nottingham
NG51PB
Velká Británie
PETNET Solutions - Mt Vemon
Lesley Harrison Building
Mount Vernon Hospital
Northwood
HA62RN
Velká Británie
PETNET Solutions - Lisses ZAC du Bois Chaland 15 rue des Pyrénées 91090 Lisses Francie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
• Další opatření k minimalizaci rizik
Držitel registračního rozhodnutí před uvedením na trh odsouhlasí v každém členském státě s narodní regulační autoritou vzdělávací program.
Držitel registračního rozhodnutí po diskuzi a souhlasu národní regulační autority v každém členském státě, ve kterém je přípravek Amyvid uváděn na trh, zajistí, že všichni lékaři, u kterých se očekává použití přípravku Amyvid, mají v okamžiku prvního uvedení přípravku na trh i později přístup ke školícímu kurzu tak, aby bylo zajištena přesná a spolehlivá interpretace PET snímků.
Školicí kurz pro lékaře by měl obsahovat tyto klíčové součásti:
• Informace o patologii amyloidu u Alzheimerovy choroby, odpovídající informace o přípravku Amyvid, jakožto indikátoru P-amyloidu včetně schválené indikace v souladu se SPC, omezení použití přípravku Amyvid, chyby v interpretaci, bezpečnostní informace a výsledky klinických studií informující o diagnostickém použití přípravku Amyvid.
• Přehled kritérií PET interpretace včetně metody posouzení snímku, kritérií interpretace a obrázky demonstrující metodologii binárního hodnocení.
• Materiál má zahrnovat demonstrační případy PET snímku za pomocí přípravku Amyvid se správnou interpretací PET snímků zkušeným hodnotitelem, každému účastníkovi by měly být poskytnuty Amyvid PET snímky pro vlastní hodnocení a pro zvýšení kvalifikace. Školení by mělo zahrnovat dostatečný počet jasně pozitivních a negativních případů, včetně nejednoznačných případů. Případy by měly být, pokud možno, histopatologicky potvrzeny.
• Musí být zajištěna odbornost a kvalifikace školitelů jak pro elektronická tak i pro osobní školení.
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU OZNAČENÍ (ŠTÍTEK) NA OCHRANNÉM OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Amyvid 800 MBq/ml injekční roztok Florbetapir-(18F)
2. OBSAH LÉČIVÉ LÁTKY
1 ml injekčního roztoku obsahuje v den a čas kalibrace (ToC) 800 MBq florbetapiru-(18F).
3. SEZNAM POMOCNÝCH LÁTEK
Bezvodý ethanol, natrium-askorbát, chlorid sodný, voda pro injekci. Pro další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 lahvička obsahuje
Objem: {Z} ml
Aktivita: {Y} MBq v {Z}ml
ToC: {DD/MM/YYYY} {hh:mm} {časová zóna}
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní použití.
Vícedávkové balení.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
^ Radioaktivní materiál
Advanced Accelerator Applications, 62660, Beuvry, Francie
|
Advanced Accelerator Applications, 01630, Saint Genis Pouilly, Francie | |
|
Advanced Accelerator Applications, 10010, Colleretto Giacosa, Itálie | |
Advanced Accelerator Applications, 86077, Pozzilli, Itálie Advanced Accelerator Applications, 47014, Meldola, Itálie Advanced Accelerator Applications, 50100, Zaragoza, Španělsko Laboratories Cyclopharma, 80440, Glisy, Francie Laboratories Cyclopharma, 31000, Toulouse, Francie PETNET Solutions, Nottingham, NG5 1PB, Velká Británie PETNET Solutions Northwood, HA6 2RN, Velká Británie PETNET Solutions, 91090, Lisses, Francie
|
8. |
POUŽITELNOST |
|
EXP: |
{DD/MM/RRRR} {hh:mm} {časová zóna} |
|
9. |
ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ |
|
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ | |
|
Veškerý nepoužitý materiál by měl být zlikvidován v souladu s místními předpisy. | |
|
11. |
NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI |
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/805/001 (10 ml) EU/1/12/805/002 (15 ml)
13. ČÍSLO ŠARŽE (A KÓD LÉČIVÉHO PŘÍPRAVKU)
Lot:
Lahvička č.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÝCH VNITŘNÍCH JEDNOTKÁCH BALENÍ OZNAČENÍ NA LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Amyvid 800 MBq/ml injekční roztok Florbetapir-(18F)
2. ZPŮSOB PODÁNÍ
Intravenózní použití
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP: ToC + 7,5 hodiny
4. ČÍSLO ŠARŽE
Lot:
Lahvička č.
5. OBSAH PODLE HMOTNOSTI, PODLE OBJEMU NEBO PODLE JEDNOTEK
< 12000 MBq v čase ToC (viz vnější obal)
6. JINÉ_
M
A Radioaktivní materiál
Advanced Accelerator Applications, 62660, Beuvry, Francie Advanced Accelerator Applications, 01630, Saint Genis Pouilly, Francie Advanced Accelerator Applications, 10010, Colleretto Giacosa, Itálie Advanced Accelerator Applications, 86077, Pozzilli, Itálie Advanced Accelerator Applications, 47014, Meldola, Itálie Advanced Accelerator Applications, 50100, Zaragoza, Španělsko Laboratories Cyclopharma, 80440, Glisy, Francie Laboratories Cyclopharma, 31000, Toulouse, Francie
PETNET Solutions, Nottingham, NG5 1PB, Velká Británie
PETNET Solutions Northwood, HA6 2RN, Velká Británie
PETNET Solutions, 91090, Lisses, Francie
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU OZNAČENÍ (ŠTÍTEK) NA OCHRANNÉM OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Amyvid 1900 MBq/ml injekční roztok Florbetapir-(18F)
2. OBSAH LÉČIVÉ LÁTKY
1 ml injekčního roztoku obsahuje v den a čas kalibrace (ToC) 1900 MBq florbetapiru-(18F).
3. SEZNAM POMOCNÝCH LÁTEK
Bezvodý ethanol, natrium-askorbát, chlorid sodný, voda pro injekci. Pro další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Injekční roztok
1 lahvička obsahuje
Objem: {Z} ml
Aktivita: {Y} MBq v {Z}ml
ToC: {DD/MM/YYYY} {hh:mm}{časová zóna}
5. ZPŮSOB A CESTA PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Intravenózní použití.
Vícedávkové balení.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
^ Radioaktivní materiál
Advanced Accelerator Applications, 62660, Beuvry, Francie
|
Advanced Accelerator Applications, 01630, Saint Genis Pouilly, Francie | |
|
Advanced Accelerator Applications, 10010, Colleretto Giacosa, Itálie | |
Advanced Accelerator Applications, 86077, Pozzilli, Itálie Advanced Accelerator Applications, 47014, Meldola, Itálie Advanced Accelerator Applications, 50100, Zaragoza, Španělsko Laboratories Cyclopharma, 80440, Glisy, Francie Laboratories Cyclopharma, 31000, Toulouse, Francie PETNET Solutions, Nottingham, NG5 1PB, Velká Británie PETNET Solutions Northwood, HA6 2RN, Velká Británie PETNET Solutions, 91090, Lisses, Francie
8. POUŽITELNOST_
EXP: {DD/MM/RRRR} {hh:mm} {časová zóna}
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý materiál by měl být zlikvidován v souladu s místními předpisy.
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Nizozemsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/805/003 (10 ml) EU/1/12/805/004 (15 ml)
13. ČÍSLO ŠARŽE (A KÓD LÉČIVÉHO PŘÍPRAVKU)
Lot:
Lahvička č.
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÝCH VNITŘNÍCH JEDNOTKÁCH BALENÍ OZNAČENÍ NA LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Amyvid 1900 MBq/ml injekční roztok Florbetapir-(18F)
2. ZPŮSOB PODÁNÍ
Intravenózní použití
Před použitím si přečtěte příbalovou informaci.
3. POUŽITELNOST
EXP: ToC + 10 hodin
4. ČÍSLO ŠARŽE
Lot:
Lahvička č.
5. OBSAH PODLE HMOTNOSTI, PODLE OBJEMU NEBO PODLE JEDNOTEK
<28500 MBq v čase ToC (viz vnější obal)
6. JINÉ_
M
A Radioaktivní materiál
Advanced Accelerator Applications, 62660, Beuvry, Francie Advanced Accelerator Applications, 01630, Saint Genis Pouilly, Francie Advanced Accelerator Applications, 10010, Colleretto Giacosa, Itálie Advanced Accelerator Applications, 86077, Pozzilli, Itálie Advanced Accelerator Applications, 47014, Meldola, Itálie Advanced Accelerator Applications, 50100, Zaragoza, Španělsko Laboratories Cyclopharma, 80440, Glisy, Francie Laboratories Cyclopharma, 31000, Toulouse, Francie
PETNET Solutions, Nottingham, NG5 1PB, Velká Británie
PETNET Solutions Northwood, HA6 2RN, Velká Británie
PETNET Solutions, 91090, Lisses, Francie
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro pacienta
AMYVID 800 MBq/ml injekční roztok AMYVID 1900 MBq/ml injekční roztok
Florbetapir-(18F)
'VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než Vám bude tento přípravek podán, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře specialisty pro nukleární medicínu, který bude řídit provádění plánovaného vyšetření.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři specialistovi pro nukleární medicínu. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Amyvid a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Amyvid podán
3. Jak se přípravek Amyvid používá
4. Možné nežádoucí účinky
5. Jak přípravek Amyvid uchovávat
6. Obsah balení a další informace
1. Co je přípravek Amyvid a k čemu se používá
Tento přípravek je radiofarmakum k použití pouze pro stanovení diagnózy.
Přípravek Amyvid obsahuje aktivní látku florbetapir-(18F).
Přípravek Amyvid se podává lidem s problémy s pamětí, aby mohl lékař provést vyšetření mozku nazývané PET snímkování. PET snímek zhotovený pomocí přípravku Amyvid pomůže Vašemu lékaři, společně s dalšími vyšetřeními funkce mozku, určit, zda máte nebo nemáte v mozku tzv. P-amyloidní plaky. Tento přípravek je určen pouze k použití u dospělých pacientů.
Výsledky testu byste měl(a) konzultovat s lékařem, který si tento test vyžádal.
Použití přípravku Amyvid je spojeno s vystavením vyšetřované osoby působení malého množství radioaktivity. Váš lékař a lékař specialista pro nukleární medicínu dospěli k závěru, že přínos tohoto vyšetření s použitím radiofarmaka převažuje nad rizikem spojeným s vystavením záření.
2. Čemu musíte věnovat pozornost, než Vám bude přípravek Amyvid podán Přípravek Amyvid nesmí být podán
- jestliže jste alergický(á) na florbetapir-(18F) nebo na kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku Amyvid sdělte lékaři specialistovi na nukleární medicínu, pokud:
- máte potíže s ledvinami
- máte potíže s játry
- jste těhotná nebo si myslíte, že byste mohla být těhotná
- pokud kojíte
Děti a dospívající
Přípravek Amyvid není určen k použití u dětí a dospívajících.
Další léčivé přípravky a přípravek Amyvid
Informujte lékaře specialistu pro nukleární medicínu o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat, protože by mohly ovlivnit hodnocení snímků.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, musíte informovat lékaře specialistu pro nukleární medicínu před podáním přípravku Amyvid. Pokud máte pochybnosti, je důležité se poradit s lékařem specialistou pro nukleární medicínu, který bude vyšetření/zákrok řídit.
Jestliže jste těhotná
Lékař specialista pro nukleární medicínu Vám tento přípravek podá v průběhu těhotenství, pouze pokud se očekává, že přínos převáží nad možnými riziky.
Jestliže kojíte
Musíte přestat kojit po dobu 24 hodin po podání injekce, a vytlačené mateřské mléko by mělo být zlikvidováno. Kojení by mělo být opět zahájeno až po dohodě s lékařem specialistou pro nukleární medicínu, který bude vyšetření řídit.
Jestliže jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se s lékařem specialistou pro nukleární medicínu ještě předtím, než vám tento přípravek bude podán.
Řízení dopravních prostředků a obsluha strojů
Možnost, že přípravek Amyvid ovlivní nepříznivě Vaši schopnost řídit nebo obsluhovat stroje, je považována za nepravděpodobnou.
Přípravek Amyvid obsahuje etanol a sodík
Tento přípravek obsahuje 10 objemových procent etanolu (alkoholu), tj. až 790 mg v jedné dávce, což odpovídá 20 ml piva nebo 8 ml vína. Toto množství může být škodlivé u osob trpících alkoholismem. Mělo by to být vzato do úvahy také u těhotných nebo kojících žen, a u osob s vysokým rizikem jako jsou pacienti s onemocněním jater nebo s epilepsií.
Tento přípravek obsahuje také natrium-askorbát a chlorid sodný. Obsah sodíku je vyšší než 1 mmol (až 37 mg v jedné dávce). To je třeba vzít v úvahu u pacientů dodržujících dietu s nízkým obsahem sodíku.
3. Jak se přípravek Amyvid používá
Pro používání přípravků obsahujících radiofarmaka, pro manipulaci s nimi a pro jejich likvidaci platí přísné předpisy. Přípravek Amyvid bude použit pouze ve speciálně kontrolovaných prostorách. S tímto přípravkem budou manipulovat a Vám jej budou podávat pouze osoby, které byly vyškoleny a mají dostatečnou kvalifikaci pro jeho bezpečné používání. Tyto osoby budou věnovat velkou pozornost bezpečnému použití tohoto přípravku a budou Vás informovat o tom, co dělají.
Dávka
Lékař specialista pro nukleární medicínu, který bude zákrok/vyšetření řídit, rozhodne, jaké množství přípravku Amyvid bude ve Vašem případě použito. Bude to nejmenší možné množství, které bude dostatečné pro získání požadovaných informací.
Obvykle doporučené množství u dospělé osoby, je 370 MBq. Megabecquerel (MBq) je jednotka, která se používá k vyjádření radioaktivity.
Podání přípravku Amyvid a provedení vyšetření/zákroku
Přípravek Amyvid se podává v injekci do Vaší žíly (intravenózní injekce). Po podání této injekce se provede propláchnutí injekce roztokem chloridu sodného, aby se zajistilo podání celé dávky.
Jedna injekce je obvykle dostatečná pro provedení vyšetření, které Váš lékař potřebuje.
Trvání vyšetření
Lékař specialista pro nukleární medicínu Vás bude informovat o obvyklém trvání vyšetření. Snímkování mozku se většinou provede přibližně 30 až 50 minut po podání injekce přípravku Amyvid.
Po podání přípravku Amyvid byste měl/a postupovat následovně:
Vyhýbejte se po dobu 24 hodin po podání injekce jakémukoli kontaktu s malými dětmi a těhotnými ženami.
Lékař specialista pro nukleární medicínu Vás bude informovat, pokud bude nutné, abyste po podání tohoto léku podnikl(a) nějaká zvláštní bezpečnostní opatření. Máte-li jakékoli otázky, obraťte se na svého lékaře specialistu pro nukleární medicínu.
Jestliže Vám bylo podání více přípravku Amyvid než mělo být podáno
Předávkování je nepravděpodobné, protože dostanete pouze jednu dávku přípravku Amyvid, kterou bude přesně kontrolovat lékař specialista pro nukleární medicínu, který bude vyšetření řídit. Nicméně v případě, že dojde k předávkování, Vám bude poskytnuto náležité ošetření. Je zejména možné, že lékař specialista pro nukleární medicínu, který bude vyšetření/zákrok řídit, může podniknout kroky, které zajistí zvýšení množství (objemu) vyloučené moče nebo stolice, aby se urychlilo odstranění radioaktivity z Vašeho těla.
Máte-li jakékoli další otázky týkající se použití přípravku Amyvid, zeptejte se prosím lékaře specialisty pro nukleární medicínu, který bude vyšetření/zákrok řídit.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Následující nežádoucí účinek přípravku Amyvid je častý (může se objevit až u 1 z 10 osob):
- bolest hlavy
Následující nežádoucí účinky přípravku Amyvid jsou málo časté (mohou se objevit až u 1 ze 100 osob).
- pocit nevolnosti (na zvracení),
- změněná chuť v ústech,
- nával horka,
- svědění,
- vyrážka, krvácení nebo bolest v místě, kde je podána injekce, nebo vyrážka v jiných místech.
Toto radiofarmakum povede k tomu, že budete vystaven(a) malému množství ionizujícího záření, které je spojeno s minimálním rizikem vzniku zhoubného nádoru nebo dědičných abnormalit (tj. genetických onemocnění). Viz také část 1.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři specialistovi pro nukleární medicínu. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Jak přípravek Amyvid uchovávat
5.
Nebude nutné, abyste tento přípravek uchovával/a. Za uchovávání tohoto přípravku je odpovědný specialista, který jej uchovává v prostorách, které jsou k tomuto účelu určeny. Uchovávání radiofarmak bude provedeno v souladu s národními právními předpisy týkajícími se radioaktivních materiálů.
Následující informace jsou určeny pouze pro odborníky.
Přípravek Amyvid nesmí být použit po uplynutí data, které je uvedeno na štítku na ochranném obalu za označením EXP: 6. Obsah balení a další informace
Co přípravek Amyvid obsahuje:
- Léčivou látkou je florbetapir-(18F).
Amyvid 1900 Mbq/ml: 1 ml injekčního roztoku obsahuje k datu a času kalibrace 1900 MBq florbetapiru-(18F).
Amyvid 800 Mbq/ml: 1 ml injekčního roztoku obsahuje k datu a času kalibrace 800 MBq florbetapiru-(18F).
- Dalšími složkami jsou bezvodý ethanol, natrium-askorbát, chlorid sodný, voda pro injekci (Viz část 2 „Přípravek Amyvid obsahuje ethanol a sodík”).
Jak přípravek Amyvid vypadá a co obsahuje toto balení
Přípravek Amyvid je čirý bezbarvý injekční roztok. Je dodáván v injekční lahvičce z borosilikátového skla (typ I) o objemu 10 ml nebo 15 ml.
Velikost balení:
Amyvid 1900 MBq/ml: Jedna vícedávková injekční lahvička o objemu 10 ml obsahující 1 až 10 ml roztoku, odpovídající 1900 až 19000 MBq k datu a času kalibrace.
Jedna vícedávková injekční lahvička o objemu 15 ml obsahující 1 až 15 ml roztoku, odpovídající 1900 až 28500 MBq k datu a času kalibrace.
Amyvid 800 MBq/ml: Jedna vícedávková injekční lahvička o objemu 10 ml obsahující 1 až 10 ml roztoku, odpovídající 800 až 8000 MBq k datu a času kalibrace.
Jedna vícedávková injekční lahvička o objemu 15 ml obsahující 1 až 15 ml roztoku, odpovídající 800 až 12000 MBq k datu a času kalibrace.
Držitel rozhodnutí o registraci:
Eli Lilly Nederland B.V., Papendorpseweg 83, 3528 BJ Utrecht, Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgique/Belgie/Belgien Eli Lilly Benelux S.A./N.V. Tél/Tel: + 32-(0)2 548 84 84 |
Lietuva Eli Lilly Holdings Limited atstovybé Tel. +370 (5) 2649600 |
|
Bt^rapnn Tn "Enn Hnan HegepnaHg" E.B. - Etnrapna Ten. + 359 2 491 41 40 |
Luxembourg/Luxemburg Eli Lilly Benelux S.A./N.V. Tél/Tel: + 32-(0)2 548 84 84 |
|
Česká republika ELI LILLY ČR, s.r.o. Tel: + 420 234 664 111 |
Magyarország Lilly Hungária Kft. Tel: + 36 1 328 5100 |
|
Danmark Eli Lilly Danmark A/S Tlf: +45 45 26 60 00 |
Malta Charles de Giorgio Ltd. Tel: + 356 25600 500 |
|
Deutschland Lilly Deutschland GmbH Tel. + 49-(0) 6172 273 2222 |
Nederland Eli Lilly Nederland B.V. Tel: + 31-(0) 30 60 25 800 |
|
Eesti Eli Lilly Holdings Limited Eesti filiaal Tel: +372 6 817 280 |
Norge Eli Lilly Norge A.S. Tlf: + 47 22 88 18 00 |
|
EkXáda OAPMAIEPB-AIAAY A.E.B.E. Tr^: +30 210 629 4600 |
Osterreich Eli Lilly Ges.m.b.H. Tel: + 43-(0) 1 711 780 |
|
Espaňa Lilly S.A. Tel: + 34-91 663 50 00 |
Polska Eli Lilly Polska Sp. z o.o. Tel: +48 (0) 22 440 33 00 |
|
France Lilly France SAS Tél: +33-(0) 1 55 49 34 34 |
Portugal Lilly Portugal Produtos Farmaceuticos, Lda Tel: + 351-21-4126600 |
|
Hrvatska Eli Lilly Hrvatska d.o.o. Tel: +385 1 2350 999 |
Románia Eli Lilly Románia S.R.L. Tel: + 40 21 4023000 |
|
Ireland Eli Lilly and Company (Ireland) Limited Tel: + 353-(0) 1 661 4377 |
Slovenija Eli Lilly farmacevtska družba, d.o.o. Tel: +386 (0)1 580 00 10 |
|
Ísland Icepharma hf. Sími + 354 540 8000 |
Slovenská republika Eli Lilly Slovakia, s.r.o. Tel: + 421 220 663 111 |
|
Italia Eli Lilly Italia S.p.A. Tel: + 39- 055 42571 |
Suomi/Finland Oy Eli Lilly Finland Ab Puh/Tel: + 358-(0) 9 85 45 250 |
Kúrcpog
Phadisco Ltd Tr|k: +357 22 715000
Sverige
Eli Lilly Sweden AB Tel: + 46-(0) 8 7378800
Latvija United Kingdom
Eli Lilly Holdings Limited párstavnieciba Latvija Eli Lilly and Company Limited Tel: +371 67364000 Tel: + 44-(0) 1256 315000
Tato příbalová informace byla naposledy schválena v {MM/RRRR}.
Podrobné informace o tomto přípravku jsou k dispozici na internetové stránce Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu/
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Úplný SPC přípravku Amyvid je přiložen v odděleném dokumentu v balení přípravku, s cílem poskytnout zdravotnickým pracovníkům další vědecké a praktické informace o podávání a používání tohoto radiofarmaka.
Prostudujte prosím přiložený SPC {SPC by měl být vložen do krabičky }.
48