Alprolix 3000 Iu
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
ALPROLIX 250 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 500 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 1000 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 2000 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 3000 IU prášek a rozpouštědlo pro injekční roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
ALPROLIX 250 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně eftrenonacogum alfa 250 IU. Po rekonstituci obsahuje jeden ml injekčního roztoku přibližně eftrenonacogum alfa 50 IU.
ALPROLIX 500 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně eftrenonacogum alfa 500 IU. Po rekonstituci obsahuje jeden ml injekčního roztoku přibližně eftrenonacogum alfa 100 IU.
ALPROLIX 1000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně eftrenonacogum alfa 1 000 IU. Po rekonstituci obsahuje jeden ml injekčního roztoku přibližně eftrenonacogum alfa 200 IU.
ALPROLIX 2000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně eftrenonacogum alfa 2 000 IU. Po rekonstituci obsahuje jeden ml injekčního roztoku přibližně eftrenonacogum alfa 400 IU.
ALPROLIX 3000 IU prášek a rozpouštědlo pro injekční roztok
Jedna injekční lahvička obsahuje nominálně eftrenonacogum alfa 3 000 IU. Po rekonstituci obsahuje jeden ml injekčního roztoku přibližně eftrenonacogum alfa 600 IU.
Účinnost (IU) se stanovuje podle Evropského lékopisu jednorázovým koagulačním testem proti vlastnímu standardu, který je uveden ve standardu pro faktor IX WHO. Specifická aktivita přípravku ALPROLIX je 55-84 IU/mg proteinu.
Eftrenonacog alfa (rekombinantní lidský koagulační faktor IX, Fc fuzní protein (rFIXFc)) obsahuje 867 aminokyselin. Je to přípravek faktoru o vysoké čistotě, který se vyrábí rekombinantní DNA technologií v buněčné linii na humánních embryonálních ledvinách (HEK) bez přídavku jakékoliv bílkoviny lidského či zvířecího původu během kultivace buněk, čištění či konečné úpravy přípravku.
Pomocná látka se známým účinkem: 0,3 mmol (6,4 mg) sodíku v injekční lahvičce. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: lyofilizovaný, bílý až téměř bílý prášek nebo koláč lyofilizátu. Rozpouštědlo: roztok je čirý až bezbarvý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a prevence krvácení u pacientů s hemofilií B (vrozený deficit faktoru IX).
Přípravek ALPROLIX lze používat ve všech věkových skupinách.
4.2 Dávkování a způsob podání
Léčba se má provádět pod dohledem lékaře, který má zkušenosti s léčbou hemofilie.
Dosud neléčení pacienti
Bezpečnost a účinnost přípravku ALPROLIX u dosud neléčených pacientů nebyly dosud stanoveny. Nejsou dostupné žádné údaje.
Monitorování léčby
V průběhu léčby je doporučeno provádět příslušné stanovení hladiny faktoru IX za účelem získání vodítka pro velikost podávané dávky i četnost opakovaných aplikací. U jednotlivých pacientů se může jejich odpověď na faktor IX lišit dosahováním různých poločasů a hodnot obnovy. Dávka podle tělesné hmotnosti může vyžadovat úpravu u pacientů s nízkou tělesnou hmotností nebo nadváhou. Zvláště u velkých chirurgických výkonů je přesné monitorování substituční léčby pomocí koagulačního vyšetření (aktivita faktoru IX v plazmě) nepostradatelné.
Při použití jednorázového vyšetření koagulace na základě in vitro tromboplastinového času (aPTT) pro stanovení aktivity faktoru IX u vzorků krve pacientů mohou být výsledky aktivity faktoru IX významně ovlivněny jak typem aPTT reagencie, tak referenčním standardem použitým v testu. To je zvláště důležité při změně laboratoře a/nebo reagencie používaného v testu.
Měření pomocí jednorázového koagulačního testu s použitím reagencie aPTT na bázi kaolinu pravděpodobně povede k podhodnocení úrovně aktivity.
Dávkování
Dávkování a trvání substituční terapie závisí na závažnosti nedostatku faktoru IX, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet podávaných jednotek rekombinantního faktoru IX Fc se vyjadřuje v mezinárodních jednotkách (International Units, IU), které jsou stanovené oproti současnému standardu WHO pro léčivé přípravky obsahující faktor IX. Aktivita FIX v plazmě se vyjadřuje buď v procentech (vzhledem k normální lidské plazmě) nebo v IU (vzhledem k mezinárodnímu standardu pro FIX v plazmě).
1 mezinárodní jednotka (IU) aktivity rekombinantního faktoru IX Fc odpovídá množství faktoru IX v 1 ml normální lidské plazmy.
Léčba on demand
Výpočet požadované dávky rekombinantního faktoru IX Fc vychází z empirického předpokladu, že podání 1 IU faktoru IX na kg tělesné hmotnosti zvýší aktivitu FIX v plazmě o 1 % normální aktivity (IU/dl). Požadovaná dávka se stanoví podle následujícího vzorce:
Požadovaný počet jednotek = tělesná hmotnost (kg) x požadovaný vzestup FIX (%) (IU/dl) x (reciproční hodnota pozorovaného recovery (IU/kg na IU/dl)}
Množství, které se má podat, a frekvence podávání mají vždy směřovat ke klinické účinnosti v individuálním případě. Pokud je nutná opakovaná dávka pro kontrolu krvácení, má se přihlížet k prodlouženému poločasu přípravku ALPROLIX (viz bod 5.2). Neočekává se zpoždění doby do dosažení maximální aktivity.
V případě následujících hemoragických příhod nemá aktivita faktoru IX během daného období klesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Tabulka 1 může posloužit jako návod pro stanovení dávky v případech krvácení a při chirurgických výkonech:
Tabulka 1: Návod pro dávkování přípravku ALPROLIX pro léčbu krvácivých příhod a chirurgických výkonů
|
Stupeň krvácení/typ chirurgického výkonu |
Požadovaná hladina FIX (%) (IU/dl) |
Frekvence dávkování (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časný hemartros, krvácení do svalů nebo do ústní dutiny |
20-40 |
Injekci opakovat každých 48 hodin, dokud se krvácení nezastaví, což se projeví ústupem bolesti nebo zahojením. |
|
Rozsáhlejší hemartros, krvácení do svalů nebo hematom |
30-60 |
Injekci opakovat každých 24 až 48 hodin, dokud bolest a akutní potíže neustoupí. |
|
Život ohrožující krvácení |
60-100 |
Injekci opakovat každých 8 až 24 hodin, dokud nepomine ohrožení. |
|
Chirurgický výkon | ||
|
Menší chirurgický výkon včetně extrakce zubu |
30-60 |
Injekci opakovat za 24 hodin, podle potřeby až do zahojení 1. |
|
Velký chirurgický výkon |
80-100 (před a po operaci) |
Injekci opakovat každých 8 až 24 hodin podle potřeby, dokud nedojde k uspokojivému zahojení rány, potom pokračovat v léčbě nejméně dalších 7 dní a udržovat aktivitu FIX mezi 30-60 % (IU/dl). |
1 U některých pacientů a za některých okolností může být interval dávkování prodloužen až na 48 hodin (viz bod 5.2 pro farmakokinetické údaje).
Profylaxe
V případě dlouhodobé profylaxe krvácení se doporučuje úvodní režim buď:
• 50 IU/kg jednou týdně, s úpravou dávky podle odpovědi pacienta nebo
• 100 IU/kg jednou za každých 10 dnů, s úpravou intervalu mezi dávkami podle odpovědi pacienta. Nejvyšší doporučená dávka pro profylaxi je 100 IU/kg.
Starší populace
Zkušenosti u pacientů ve věku >65 let jsou omezené.
Pediatrická populace
U dětí ve věku do 12 let mohou být nutné vyšší nebo častější dávky a doporučená úvodní dávka je 50-60 IU/kg každých 7 dnů. U dospívajících ve věku 12 let a starších platí stejná doporučení jako u dospělých. Viz body 5.1 a 5.2.
Nejvyšší doporučená dávka pro profylaxi je 100 IU/kg.
Způsob podání Intravenózní podání.
V případě podání přípravku pacientem nebo pečovatelem je nutné příslušné proškolení.
Přípravek ALPROLIX má být podáván intravenózně injekcí během několika minut. Rychlost podávání má být stanovena podle úrovně pohodlí pacienta a nemá přesáhnout 10 ml/min.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku (rekombinantní lidský koagulační faktor IX a/nebo Fc doménu) nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ALPROLIX jsou možné reakce z přecitlivělosti alergického typu. Pokud se objeví příznaky hypersenzitivity, pacienti mají být poučeni, aby okamžitě ukončili používání léčivého přípravku a kontaktovali svého lékaře. Pacienti mají být informováni o časných známkách reakcí z přecitlivělosti, jako je kopřivka, generalizovaná kopřivka, tlak na hrudi, sípání, hypotenze a anafylaxe.
V případě anafylaktického šoku je nutné dodržovat všeobecné lékařské postupy pro léčbu šoku.
Inhibitory
Po opakované léčbě přípravky obsahujícími lidský koagulační faktor IX mají být pacienti monitorováni s ohledem na výskyt neutralizačních protilátek (inhibitorů), které mají být kvantifikovány v Bethesda jednotkách (BU) pomocí vhodného biologického testu.
V literatuře byly zaznamenány údaje prokazující korelaci mezi výskytem inhibitoru faktoru IX a alergickými reakcemi. Pacienti s alergickými reakcemi proto mají být vyšetřeni na přítomnost inhibitoru.
Je třeba mít na paměti, že pacienti s inhibitory faktoru IX mohou mít zvýšené riziko anafylaxe při následné expozici faktoru IX.
Z důvodu rizika alergických reakcí na přípravky obsahující faktor IX má být úvodní podání faktoru IX prováděno podle úsudku lékaře a pod dohledem lékaře při zajištění možnosti náležité léčby alergických reakcí.
Tromboembolismus
Z důvodu možného rizika trombotických komplikací u přípravků obsahujících faktor IX má být zahájeno klinické sledování výskytu časných známek trombotické a konsumpční koagulopatie s vhodným biologickým testováním při podávání tohoto přípravku pacientům s onemocněním jater, po operaci, novorozencům nebo pacientům s rizikem trombotické příhody nebo diseminované intravaskulární koagulace (DIK). Přínos léčby přípravkem ALPROLIX v těchto situacích má být zvažován oproti riziku těchto komplikací.
Kardiovaskulární příhody
U pacientů se stávajícími kardiovaskulárními rizikovými faktory může substituční léčba pomocí FIX zvýšit kardiovaskulární riziko.
Komplikace související s použitím katétru
Pokud je nutné použití centrálního žilního katétru (CŽK), je nutno zvážit riziko komplikací spojené s jeho použitím, včetně lokálních infekcí, bakteriémie a trombózy v místě zavedení katétru.
Záznam čísla šarže
Důrazně se doporučuje, aby vždy, když je přípravek ALPROLIX podáván pacientovi, byl zaznamenán název a číslo šarže přípravku, aby byla jasná souvislost mezi pacientem a šarží léčivého přípravku.
Pediatrická populace
Uvedená upozornění a preventivní opatření platí pro dospělé i děti.
Upozornění na pomocné látky
Tento léčivý přípravek obsahuje 0,3 mmol (nebo 6,4 mg) sodíku v injekční lahvičce. Nutno vzít v úvahu u pacientů na dietě s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Nebyly hlášeny žádné interakce přípravku ALPROLIX s jinými léčivými přípravky. Nebyly provedeny žádné studie interakcí.
4.6 Fertilita, těhotenství a kojení
Těhotenství a kojení
S přípravkem ALPROLIX se neprováděly reprodukční studie u zvířat. Byla provedena studie placentárního přenosu u myší (viz bod 5.3). Vzhledem k vzácnému výskytu hemofilie B u žen nejsou k dispozici zkušenosti týkající se použití faktoru IX během těhotenství a kojení. Proto má být faktor IX používán během těhotenství a kojení pouze tehdy, pokud je to jednoznačně indikováno.
Fertilita
Nejsou k dispozici žádné údaje o fertilitě. U zvířat nebyly provedeny žádné studie fertility s přípravkem ALPROLIX.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek ALPROLIX nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Vzácně byly pozorovány hypersenzitivita nebo alergické reakce (které mohou zahrnovat angioedém, pálení a píchání v místě podání infuze, třesavku, zrudnutí, generalizovanou kopřivku, bolest hlavy, kopřivku, hypotenzi, letargii, nauzeu, neklid, tachykardii, tíseň na hrudi, brnění, zvracení, sípání), které mohou v některých případech vyústit v těžkou anafylaxi (včetně šoku). V některých případech se tyto reakce vyvinuly v závažnou anafylaxi a objevily se v úzké časové souvislosti s objevením inhibitorů faktoru IX (viz také bod 4.4). Byl hlášen nefrotický syndrom po pokusu o navození imunitní tolerance u pacientů s hemofilií B s inhibitory faktoru IX a anamnézou alergické reakce.
U pacientů s hemofilií B se mohou vytvořit neutralizační protilátky (inhibitory) proti faktoru IX. Pokud se takové inhibitory objeví, projeví se jako nedostatečná klinická odpověď. V takových případech se doporučuje vyhledat specializované pracoviště k léčbě hemofilie.
Existuje možné riziko tromboembolických epizod po podání přípravků s obsahem faktoru IX, přičemž toto riziko je vyšší u přípravků s nižší čistotou. Používání přípravků s obsahem faktoru IX s nízkou čistotou bylo spojeno s případy infarktu myokardu, diseminované intravaskulární koagulace, žilní trombózy a plicní embolizace. Používání faktoru IX s vysokou čistotou vzácně souviselo s tromboembolickými komplikacemi.
Tabulkový seznam nežádoucích účinků
Frekvence v tabulce uvedené níže byly pozorovány u celkem 153 pacientů se závažnou hemofilií B v klinických studiích fáze III a v prodloužené studii. Celkový počet dnů expozice byl 17 080 s mediánem 100 (rozmezí 1-351) dnů expozice na pacienta.
Tabulka 2 uvedená níže vychází z klasifikace orgánových systémů MedDRA (SOC a preferovaná úroveň termínu).
Frekvence byly hodnoceny podle následující konvence: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000), není známo (z dostupných údajů nelze určit).
V rámci každé skupiny frekvence jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Třídy orgánových systémů dle MedDRA |
Nežádoucí účinky |
Kategorie dle frekvence |
|
Poruchy metabolismu a výživy |
Snížená chuť k jídlu |
Méně časté |
|
Poruchy nervového systému |
Závrať Dysgeuzie |
Časté Méně časté Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Orální parestézie Zápach z úst |
Časté Méně časté |
|
Poruchy ledvin a močových cest |
Obstrukční uropatie Hematurie Renální kolika |
Časté Méně časté Méně časté |
|
Celkové poruchy a reakce v místě aplikace |
Únava Bolest v místě podání infuze |
Méně časté Méně časté |
Pediatrická populace
Očekává se, že frekvence, typ a závažnost nežádoucích účinků u dětí bude podobná jako u dospělých. Rozsah a věková charakteristika bezpečnostní databáze u dětí je uvedena v bodě 5.1
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Účinky vyšších než doporučených dávek přípravku ALPROLIX nebyly popsány.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Hemostatika, koagulační faktor IX, ATC kód: B02BD04 Mechanismus účinku
Faktor IX je glykoprotein s jedním řetězcem s molekulovou hmotností přibližně 68 000 daltonů. Je to koagulační faktor závislý na vitaminu K. Faktor IX je aktivován faktorem XIa ve vnitřním systému koagulační kaskády a komplexem faktoru VII/tkáňového faktoru ve vnějším systému. Aktivovaný faktor IX v kombinaci s aktivovaným faktorem VIII aktivuje faktor X. Aktivovaný faktor X transformuje protrombin na trombin. Trombin následně transformuje fibrinogen na fibrin a dochází k vytvoření sraženiny. Hemofilie B je pohlavně vázaná dědičná porucha srážlivosti krve způsobená sníženou hladinou faktoru IX, v důsledku které dochází ke krvácení do kloubů, svalů nebo vnitřních orgánů, buď spontánnímu, nebo jako následek úrazu při nehodě či chirurgickém zákroku. Substituční léčbou se hladina faktoru IX v plazmě zvýší, díky čemuž je možná přechodná korekce deficitu faktoru a korekce sklonu ke krvácení.
Přípravek ALPROLIX (eftrenonacog alfa) je dlouhodobě působící, plně rekombinantní fuzní protein, který se skládá z lidského koagulačního faktoru IX kovalentně vázaného na Fc doménu lidského imunoglobulinu G1 a je vyráběn rekombinantní DNA technologií.
Fc region lidského imunoglobulinu G1 se váže na neonatální Fc receptor. Tento receptor je exprimován v průběhu života jako součást přirozeně se vyskytujícího mechanismu, který chrání imunoglobuliny před lysozomální degradací navrácením těchto proteinů zpět do oběhu, což vede k jejich dlouhému plazmatickému biologickému poločasu.
Klinická účinnost a bezpečnost
Bezpečnost, účinnost a farmakokinetika přípravku ALPROLIX byly hodnoceny ve 2 mezinárodních, otevřených, pivotních studiích - studii fáze 3 označené jako studie I a pediatrické studii fáze 3 označené jako studie II (viz Pediatrická populace).
Studie I srovnávala účinnost každého ze 2 profylaktických režimů léčby (fixní týdenní interval a individualizovaný interval) pro léčbu on demand. Do studie bylo zařazeno celkem 123 dříve léčených mužských pacientů (12 až 71 let) se závažnou hemofilií B (<2 % aktivity endogenního FIX). Všichni pacienti byli léčeni přípravkem ALPROLIX a byli sledováni po dobu až 77 týdnů.
V ramenu fixního týdenního intervalu dostávali pacienti přípravek ALPROLIX v rámci běžné profylaxe od dávky 50 IU/kg. V ramenu individualizovaného intervalu dostávali pacienti přípravek ALPROLIX v rámci běžné profylaxe ve fixní dávce 100 IU/kg v intervalu dávkování od podání jednou za 10 dnů. Kromě toho byla ve studii I hodnocena hemostatická účinnost při léčbě epizod krvácení a stanovena hemostatická účinnost během perioperativní léčby u pacientů podstupujících velké chirurgické výkony.
Profylaxe ve _fixních týdenních a individualizovaných intervalech:
U hodnotitelných pacientů zařazených do profylaktického ramena s fixním týdenním intervalem ve studii I byl medián týdenní dávky 45,17 IU/kg (interkvartilní rozmezí 38,1-53,7). U hodnotitelných pacientů zařazených do profylaktického ramena s individualizovaným intervalem ve studii I byl medián intervalu podávání 12,53 dnů (interkvartilní rozmezí 10,4-13,4).
U pacientů hodnotitelných z hlediska účinnosti byl medián četnosti krvácení přepočtené na jeden rok (Annualised Bleeding Rates, ABR) 2,95 (interkvartilní rozmezí (1,01-4,35) u pacientů s profylaxí s fixním týdenním intervalem, 1,38 (interkvartilní rozmezí (0,00-3,43) u pacientů s individualizovaným intervalem a 17,69 (interkvartilní rozmezí 10,77-23,24) u pacientů léčených podle potřeby. Žádné epizody krvácení se nevyskytly u 42 % pacientů léčených v režimu individualizované profylaxe a u 23,0 % pacientů léčených v režimu týdenní profylaxe. Podíl pacientů s >1 cílovým kloubem při výchozím stavu ve skupině s individualizovaným intervalem profylaxe byl nižší než ve skupině s týdenní profylaxí (27,6 % a 57,1 %, v uvedeném pořadí).
Je třeba uvést, že ABR není srovnatelný mezi různými koncentráty faktoru a mezi různými klinickými studiemi.
Léčba krvácení: Ze 636 krvácivých příhod pozorovaných během studie I bylo 90,4 % zvládnuto pomocí 1 injekce a celkem 97,3 % pomocí 2 nebo méně injekcí. Medián průměrné dávky na injekci pro léčbu epizody krvácení byl 46,07 (interkvartilní rozmezí 32,86-57,03) IU/kg. Medián celkové dávky pro léčbu epizody krvácení byl 51,47 IU/kg (interkvartilní rozmezí 35,21-61,73) v ramenu týdenní profylaxe,
49,62 IU/kg (35,71-94,82) v ramenu profylaktického individualizovaného intervalu a 46,58 IU/kg (33,33-59,41) v ramenu léčby podle potřeby.
Pediatrická populace
Do studie II bylo zařazeno celkem 30 dříve léčených pediatrických pacientů mužského pohlaví se závažnou hemofilií B (<2 % aktivity endogenního FIX). Pacienti byli ve věku do 12 let (15 bylo ve věku <6 let a 15 bylo ve věku 6 až <12 let). Všichni pacienti byli léčeni přípravkem ALPROLIX a byli sledováni po dobu až 52 týdnů.
Všech 30 pacientů bylo léčeno přípravkem ALPROLIX v režimu individualizované profylaktické dávky počínaje 50-60 IU/kg každých 7 dnů s úpravou dávky na maximální dávku 100 IU/kg a intervalem podávání minimálně jednou týdně a maximálně dvakrát týdně.
Režim individualizované profylaxe:
Medián průměrné týdenní dávky přípravku ALPROLIX byl 59,40 IU/kg a (interkvartilní rozmezí 52,95 až 64,78 IU/kg) u pacientů ve věku <6 let a 57,78 IU/kg (interkvartilní rozmezí 51,67 až 65,01 IU/kg) u pacientů ve věku 6 až <12 let. Medián intervalu podávání byl celkově 6,99 dne (interkvartilní rozmezí 6,94 až 7,03) bez rozdílu ve středním intervalu dávkování mezi věkovými kohortami. S výjimkou jednoho pacienta, jehož poslední předepsaná dávka byla 100 IU/kg každých 5 dnů, byly u dalších 29 pacientů poslední předepsané dávky až 70 IU/kg každých 7 dnů. U 33 % pediatrických pacientů nebyly zaznamenány žádné epizody krvácení.
Medián četnosti krvácení přepočtené na jeden rok u pacientů ve věku <12 let hodnotitelných z hlediska účinnosti byl 1,97 (interkvartilní rozmezí 0,00-3,13).
Léčba epizod krvácení: Ze 60 krvácivých příhod pozorovaných během studie II bylo 75 % zvládnuto pomocí 1 injekce a celkem 91,7 % pomocí 2 nebo méně injekcí. Medián průměrné dávky na injekci pro léčbu epizody krvácení byl 63,51 (interkvartilní rozmezí 48,92-99,44) IU/kg. Medián celkové dávky pro léčbu epizody krvácení byl 68,22 IU/kg (interkvartilní rozmezí 50,89-126,19).
Perioperační léčba (chirurgická profylaxe):
Ve studii I a v prodloužené studii bylo provedeno a vyhodnoceno celkem 29 velkých chirurgických výkonů u 19 pacientů (17 dospělých, 1 dospívající a 1 pediatrický pacient ve věku <12 let). Z 29 velkých chirurgických výkonů vyžadovalo 24 chirurgických výkonů (82,8 %) jednu předoperační dávku pro udržení hemostázy během operace. Medián průměrné dávky na injekci pro udržení hemostázy během operace byl 94,7 IU/kg (rozmezí 49 až 152 IU/kg). Celková dávka v den operace se pohybovala od 51 do 318 IU/kg a celková dávka ve 14denní pooperační fázi se pohybovala od 60 do 1 947 IU/kg.
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s přípravkem ALPROLIX byly provedeny u dříve léčených pacientů se závažnou hemofilií B. Údaje prezentované v tomto bodě byly získány v jednorázových testech srážlivosti s použitím reagencie aPTT na bázi křemíku kalibrovaného proti plazmatickým standardům faktoru IX.
Farmakokinetické vlastnosti byly hodnoceny u 22 pacientů (>19 let) léčených přípravkem ALPROLIX (rFIXFc). Po období bez léčby v trvání minimálně 120 hodin (5 dnů) dostali pacienti jednotlivou dávku 50 IU/kg přípravku ALPROLIX. Farmakokinetické vzorky byly shromážděny před dávkou a pak následně v 11 časových bodech až do 240 hodin (10 dnů) po podání dávky. Farmakokinetické parametry non-kompartmentové analýzy po podání dávky 50 IU/kg přípravku ALPROLIX jsou uvedeny v tabulce 3.
|
Farmakokinetické parametry1 |
ALPROLIX (95% IS) |
|
N=22 | |
|
Přírůstková recovery (IU/dl na IU/kg) |
0,92 (0,77-1,10) |
|
AUC/Dávka |
31,58 |
|
(IU*h/dl na IU/kg) |
(28,46-35,05) |
|
Cmax (IU/dl) |
46,10 (38,56-55,11) |
|
CL (ml/h/kg) |
3,17 (2,85-3,51) |
|
t/ (h) |
77,60 (70,05-85,95) |
|
t/a (h)2 |
5,03 (3,20-7,89) |
|
t/2p (h)2 |
82,12 (71,39-94,46) |
|
MRT (h) |
95,82 (88,44-106,21) |
|
Vss (ml/kg) |
303,4 (275,1-334,6) |
|
Čas do 1 % (dny)2 |
11,22 (10,20-12,35) |
1 Farmakokinetické parametry jsou uvedeny jako geometrický průměr (95% IS)
2 Tyto farmakokinetické parametry byly získány kompartmentovou analýzou
Zkratky: IS = interval spolehlivosti; Cmax= maximální aktivita; AUC = plocha pod křivkou aktivita FIX - čas; t>/2a = distribuční poločas; t/p = eliminační poločas;t/= terminální biologický poločas; CL = clearance; Vss = distribuční objem v ustáleném stavu; MRT = průměrný rezidenční čas.
Eliminační poločas přípravku ALPROLIX (82 hodin) je ovlivněn Fc oblastí, u které bylo na zvířecích modelech prokázáno, že je řízena cyklickými mechanismy neonatálního FC receptoru.
Na základě údajů o aktivitě FIX od 161 pacientů všech věkových skupin (2-76 let) s tělesnou hmotností od 12,5 kg do 186,7 kg ve třech klinických studiích (12 pacientů ve studii fáze 1/2a, 123 pacientů ve studii fáze I a 26 pacientů ve studii fáze II) byl vytvořen populační farmakokinetický model. Odhad CL přípravku ALPROLIX pro typického dospělého pacienta s tělesnou hmotností 70 kg je 2,30 dl/h a distribuční objem přípravku ALPROLIX v ustáleném stavu je 194,8 dl. Pozorovaný profil průměrné (SD) doby aktivity po jedné dávce přípravku ALPROLIX u pacientů s těžkou hemofilií B je uveden níže (viz tabulka 4).
Tabulka 4: Pozorovaná průměrná (SD) aktivita FIX [IU/dl] po jedné dávce přípravku ALPROLIX1 u
|
pacientů ve vě |
ku >12 |
et | ||||||||||
|
Dávka (IU/kg) |
10 minut |
1 h |
3 h |
6 h |
24 h |
48 h |
96 h |
144 h |
168 h |
192 h |
240 h |
288 h |
|
50 |
52,9 |
34,5 |
28,7 |
25,1 |
15,1 |
9,7 |
5,0 |
3,4 |
3,2 |
2,6 |
2,1 |
NA |
|
(30,6) |
(7,3) |
(6,7) |
(5,1) |
(3,9) |
(3,0) |
(1,6) |
(1,1) |
(1,9) |
(1,0) |
(0,9) | ||
|
100 |
112 |
NA |
77,1 |
NA |
36,7 |
21,8 |
10,1 |
NA |
4,81 |
NA |
2,86 |
2,30 |
|
(24) |
(12,8) |
(8,0) |
(4,8) |
(2,6) |
(1,67) |
(0,98) |
(0,94) | |||||
Pediatrická populace
Farmakokinetické parametry přípravku ALPROLIX byly stanoveny pro dospívající ve studii I (odběr vzorků na farmakokinetické vyšetření byl proveden před podáním dávky s následným hodnocením při více časových bodech až do 336 hodin (14 dnů) po podání dávky) a u dětí ve studii II (odběr vzorků na farmakokinetické vyšetření byl proveden před podáním dávky s následným hodnocením při 7 časových bodech až do 168 hodin (7 dnů) po podání dávky). Tabulka 5 uvádí farmakokinetické parametry vypočtené z pediatrických údajů u 35 pacientů ve věku do 18 let.
Tabulka 5: Srovnání FK parametrů přípravku ALPROLIX (rFIXFc) podle věkové kategorie
|
FK parametry1 |
Studie II |
Studie I | |
|
<6 let (2, 4) |
6 až <12 let (6, 10) |
12 až <18 let (12, 17) | |
|
N = 11 |
N = 13 |
N = 11 | |
|
IR (IU/dl na IU/kg) |
0,5989 (0,5152; 0,6752) |
0,7170 (0,6115; 0,8407) |
0,8470 (0,6767; 1,0600) |
|
AUC/dávka (IU*h/dl na IU/kg) |
22,71 (20,32; 25,38) |
28,53 (24,47; 33,27) |
29,50 (25,13; 34,63) |
|
t/ (h) |
66,49 (55,86; 79,14) |
70,34 (60,95; 81,17) |
82,22 (72,30; 93,50) |
|
MRT (h) |
83,65 (71,76; 97,51) |
82,46 (72,65; 93,60) |
93,46 (81,77; 106,81) |
|
CL (ml/h/kg) |
4,365 (3,901; 4,885) |
3,505 (3,006; 4,087) |
3,390 (2,888; 3,979) |
|
Vss (ml/kg) |
365,1 (316,2; 421,6) |
289,0 (236,7; 352,9) |
316,8 (267,4; 375,5) |
1 Farmakokinetické parametry odvozené z non-kompartmentové analýzy jsou uvedeny jako geometrický průměr (95% IS) Zkratky: IS = interval spolehlivosti; IR = přírůstková recovery; AUC = plocha pod křivkou aktivita FIX - čas; t>/2 = terminální biologický poločas;
MRT = střední rezidenční čas; CL = clearance; Vss = distribuční objem v ustáleném stavu
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě testu trombogenicity u králíků (Wesslerův model stázy) a studií toxicity po opakovaném podávání (které zahrnovaly hodnocení lokální toxicity, samčích reprodukčních orgánů a elektrokardiografické parametry) u potkanů a opic neodhalily žádné zvláštní riziko pro člověka. Studie hodnotící genotoxicitu, kancerogenní potenciál, reprodukční toxicitu nebo embryofetální vývoj nebyly provedeny. Ve studii placentárního přenosu bylo prokázáno, že přípravek ALPROLIX prochází u myší placentou v malých množstvích.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek Sacharosa Histidin Mannitol Polysorbát 20
Hydroxid sodný (pro úpravu pH)
Kyselina chlorovodíková (pro úpravu pH)
Rozpouštědlo Chlorid sodný Voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky.
Je třeba používat pouze dodávanou infuzní soupravu, protože v důsledku adsorpce lidského koagulačního faktoru IX na vnitřní povrch některých injekčních zařízení může dojít k selhání léčby.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 4 roky
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 30°C) po jedno nepřetržité období nepřesahující 6 měsíců. Jakmile byl přípravek jednou vyjmut z chladničky, je nutné na obalu vyznačit datum, kdy k tomu došlo. Po uchovávání při pokojové teplotě nesmí být přípravek vrácen do chladničky. Přípravek se nesmí používat po uplynutí doby použitelnosti uvedené na injekční lahvičce nebo šest měsíců po vyjmutí vnějšího obalu z chladničky, cokoli nastane jako první.
Po rekonstituci
Chemická a fyzikální stabilita byla prokázána po dobu 6 hodin, když je přípravek uchováván při pokojové teplotě (až do 30 °C). Přípravek musí být zlikvidován, pokud není použit během 6 hodin.
Z mikrobiologického hlediska by měl být přípravek použit okamžitě po rekonstituci. Není-li použit okamžitě, přechází zodpovědnost za dobu a podmínky uchovávání přípravku po otevření a před použitím na uživatele. Chraňte před přímým slunečním světlem.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení a zvláštní vybavení pro použití a podání
Jedno balení obsahuje:
- prášek v injekční lahvičce (sklo třídy 1) s chlorbutylovou pryžovou zátkou,
- 5 ml rozpouštědla v předplněné injekční stříkačce (sklo třídy 1) s brombutylovou pryžovou pístovou zátkou,
- nástavec pístu,
- sterilní adaptér injekční lahvičky pro rekonstituci,
- sterilní infuzní souprava,
- tampón(y) napuštěné alkoholem,
- náplast(i),
- gázový(é) polštářek(ky).
Balení po 1.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Prášek na injekci v každé injekční lahvičce musí být rekonstituován dodávaným rozpouštědlem (roztok chloridu sodného) z předplněné injekční stříkačky pomocí sterilního adaptéru injekční lahvičky na rekonstituci.
S injekční lahvičkou se má jemně kroužit, dokud se všechen prášek nerozpustí.
Pro další informace o rekonstituci a podání si přečtěte příbalovou informaci.
Rekonstituovaný roztok má být čirý až mírně opalescentní a bezbarvý. Rekonstituovaný léčivý přípravek je třeba před podáním zkontrolovat zrakem, zda neobsahuje částečky hmoty a nedošlo ke změně barvy. Nepoužívejte roztoky, které jsou zakalené nebo které obsahují usazeninu.
Tento přípravek je určen pouze pro jedno použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Biogen Idec Ltd Innovation House 70 Norden Road Maidenhead Berkshire SL64AY Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/16/1098/001
EU/1/16/1098/002
EU/1/16/1098/003
EU/1/16/1098/004
EU/1/16/1098/005
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace:
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE/VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce biologické léčivé látky
Biogen Inc
5000 Davis Drive
Research Triangle Park
North Carolina
27709-4627
USA
Název a adresa výrobce odpovědného za propouštění šarží
Swedish Orphan Biovitrum AB (publ)
Strandbergsgatan 49 11276 Stockholm Švédsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU_
ALPROLIX 250 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 500 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 1000 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 2000 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 3000 IU prášek a rozpouštědlo pro injekční roztok eftrenonacogum alfa
rekombinantní koagulační faktor IX, Fc fuzní protein
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK_
Prášek: Eftrenonacogum alfa 250 IU(přibližně 50 IU/ml po rekonstituci) Prášek: Eftrenonacogum alfa 500 IU (přibližně 100 IU/ml po rekonstituci) Prášek: Eftrenonacogum alfa 1000 IU (přibližně 200 IU/ml po rekonstituci) Prášek: Eftrenonacogum alfa 2000 IU (přibližně 400 IU/ml po rekonstituci) Prášek: Eftrenonacogum alfa 3000 IU (přibližně 600 IU/ml po rekonstituci)
3. SEZNAM POMOCNÝCH LÁTEK
Prášek: sacharosa, histidin, mannitol, polysorbát 20, hydroxid sodný, kyselina chlorovodíková
Rozpouštědlo: chlorid sodný voda na injekci
4. LÉKOVÁ FORMA A OBSAH BALENÍ_
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s práškem, 5 ml rozpouštědla v předplněné injekční stříkačce, 1 nástavec pístu, 1 adaptér injekční lahvičky, 1 infuzní souprava, 2 alkoholové tampóny, 2 náplasti, 1 gáza
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Intravenózní podání, po rekonstituci.
Před použitím si přečtěte příbalovou informaci.
Instrukční video, jak připravit a podat přípravek ALPROLIX, je k dispozici po oskenování QR kódu chytrým telefonem nebo na webových stránkách
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
Spotřebujte během 6 hodin po rekonstituci.
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Uchovávejte v chladničce.
Chraňte před mrazem.
Může být uchováváno při pokojové teplotě (do 30°C) po jedno nepřetržité období nepřesahující 6 měsíců. Po uchovávání při pokojové teplotě nesmí být přípravek vrácen do chladničky.
Datum vyjmutí z chladničky:
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Biogen Idec Limited
Innovation House, 70 Norden Road, Maidenhead, Berkshire, SL6 4AY,Velká Británie
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/16/1098/001 EU/1/16/1098/002 EU/1/16/1098/003 EU/1/16/1098/004 EU/1/16/1098/005
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ALPROLIX 250 ALPROLIX 500 ALPROLIX 1000 ALPROLIX 2000 ALPROLIX 3000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ALPROLIX 250 IU prášek na injekci
ALPROLIX 500 IU prášek na injekci
ALPROLIX 1000 IU prášek na injekci
ALPROLIX 2000 IU prášek na injekci
ALPROLIX 3000 IU prášek na injekci
eftrenonacogum alfa rekombinantní koagulační faktor IX i.v.
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU 500 IU 1000IU 2000 IU 3000IU
6. JINÉ
25
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
Rozpouštědlo pro přípravek ALPROLIX chlorid sodný voda na injekci
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
5 ml
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
ALPROLIX 250 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 500 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 1000 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 2000 IU prášek a rozpouštědlo pro injekční roztok ALPROLIX 3000 IU prášek a rozpouštědlo pro injekční roztok
eftrenonacogum alfa
rekombinantní koagulační faktor IX, Fc fuzní protein
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky, je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
• Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
• Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
• Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
• Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek ALPROLIX a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek ALPROLIX používat
3. Jak se přípravek ALPROLIX používá
4. Možné nežádoucí účinky
5. Jak přípravek ALPROLIX uchovávat
6. Obsah balení a další informace
7. Instrukce pro přípravu a podávání
1. Co je přípravek ALPROLIX a k čemu se používá
Přípravek ALPROLIX obsahuje léčivou látku eftrenonacog alfa, rekombinantní koagulační faktor IX a Fc fuzní protein. Faktor IX je bílkovina, která se přirozeně vytváří v těle a je nutná pro srážení krve a zastavení krvácení.
Přípravek ALPROLIX je lék používaný pro léčbu a prevenci krvácení u všech věkových skupin pacientů s hemofilií B (dědičná porucha krvácení způsobená nedostatkem faktoru IX).
Přípravek ALPROLIX se připravuje rekombinantní technologií bez přidání jakýchkoli lidských nebo zvířecích složek ve výrobním procesu.
Jak přípravek ALPROLIX účinkuje
Pacientům s hemofilií B chybí faktor IX nebo neúčinkuje správně. Přípravek ALPROLIX se používá pro náhradu chybějícího nebo defektního faktoru IX. Přípravek ALPROLIX zvyšuje hladinu faktoru IX v krvi a dočasně upravuje sklon ke krvácení. Fc fuzní protein v přípravku ALPROLIX prodlužuje dobu jeho účinnosti.
2. Čemu musíte věnovat pozornost, než začnete přípravek ALPROLIX používat Nepoužívejte přípravek ALPROLIX:
• jestliže jste alergický(á) na eftrenonacog alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před použitím přípravku ALPROLIX se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
• Existuje malá možnost, že se u Vás může objevit anafylaktická reakce (závažná, náhlá alergická reakce) na přípravek ALPROLIX. Známky alergických reakcí mohou zahrnovat svědění kůže celého těla, kopřivku, tlak na hrudi, problémy s dýcháním a nízký krevní tlak. Pokud se některé z těchto příznaků objeví, ukončete ihned podávání injekce a kontaktujte svého lékaře.
• Informujte svého lékaře, pokud si myslíte, že Vaše krvácení není pod kontrolou pomocí dávky, kterou dostáváte, protože pro to může být několik důvodů. Například tvorba protilátek (také známých jako inhibitory) proti faktoru IX je známá komplikace, která může během léčby hemofilie B nastat. Protilátky zabraňují správnému účinku léčby. To by měl zkontrolovat Váš lékař. Nezvyšujte celkovou dávku přípravku ALPROLIX určenou pro kontrolu Vašeho krvácení bez konzultace se svým lékařem.
Pacienti s inhibitorem faktoru IX mohou mít zvýšené riziko anafylaxe při léčbě faktorem IX v budoucnu. Proto pokud se u Vás objeví alergické reakce, které jsou popsány výše, měl(a) byste být vyšetřen(a) na přítomnost inhibitoru.
Přípravky obsahující faktor IX mohou zvyšovat riziko nežádoucích krevních sraženin v krvi, zvláště pokud jsou u Vás přítomny rizikové faktory rozvoje krevních sraženin. Příznaky možné nežádoucí krevní sraženiny mohou zahrnovat: bolest a/nebo citlivost podél průběhu žíly, neočekávaný otok horní nebo dolní končetiny nebo náhlou dušnost či problémy s dýcháním.
Komplikace související s použitím katétru
Pokud je nutné, abyste používal(a) centrální žilní katétr (CŽK), mělo by se zvážit riziko komplikací spojené s jeho použitím včetně lokálních infekcí, bakteriémie a krevních sraženin v místě zavedení katétru.
Dokumentace
Důrazně se doporučuje, abyste při každém podání přípravku ALPROLIX zaznamenal(a) název a číslo šarže přípravku.
Další léčivé přípravky a přípravek ALPROLIX
Informujte svého lékaře nebo lékárníka o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Řízení dopravních prostředků a obsluha strojů
Přípravek ALPROLIX nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek ALPROLIX obsahuje sodík
Tento přípravek obsahuje 6,4 mg sodíku v injekční lahvičce po přípravě. Informujte svého lékaře, pokud jste na dietě s nízkým obsahem sodíku.
3. Jak se přípravek ALPROLIX používá
Léčba přípravkem ALPROLIX má být zahájena lékařem, který má zkušenosti s léčbou
pacientů s hemofilií. Vždy používejte tento přípravek přesně podle pokynů svého lékaře (viz bod 7). Pokud
si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Přípravek ALPROLIX se podává injekcí do žíly. Vy nebo někdo jiný můžete aplikovat přípravek ALPROLIX po příslušném proškolení. Váš lékař se rozhodne, jakou dávku přípravku ALPROLIX (v mezinárodních jednotkách neboli IU) budete dostávat. Tato dávka bude záviset na Vašich individuálních potřebách substituční léčby faktorem IX a na tom, zda se používá pro prevenci nebo léčbu krvácení. Informujte svého lékaře, pokud se domníváte, že Vaše krvácení není pod kontrolou pomocí dávky, kterou dostáváte.
To, jak často budete potřebovat injekci, bude záviset na tom, jak dobře u Vás přípravek ALPROLIX účinkuje. Váš lékař provede příslušné laboratorní testy, aby se ujistil, že máte odpovídající hladinu faktoru IX v krvi.
Léčba krvácení
Dávka přípravku ALPROLIX se vypočte podle Vaší tělesné hmotnosti a hladiny faktoru IX, které má být dosaženo. Cílová hladina faktoru IX bude záviset na závažnosti a lokalizaci krvácení.
Prevence krvácení
Pokud používáte přípravek ALPROLIX pro prevenci krvácení, vypočte Vám dávku Váš lékař.
Obvyklá dávka přípravku ALPROLIX je 50 IU na kg tělesné hmotnosti podávaných jednou týdně nebo 100 IU na kg tělesné hmotnosti podávaných každých 10 dnů. Dávka nebo interval mohou být upraveny Vaším lékařem. V některých případech, zejména u mladších pacientů, mohou být nutné kratší intervaly mezi dávkami nebo vyšší dávky.
Použití u dětí a dospívajících
Přípravek ALPROLIX se může používat u dětí a dospívajících každého věku. U dětí ve věku do 12 let mohou být nutné vyšší dávky nebo častější injekce a obvyklá dávka je 50 až 60 IU na kg tělesné hmotnosti podávaných jednou za 7 dnů.
Jestliže jste použil(a) více přípravku ALPROLIX, než jste měl(a)
Informujte co možná nejdříve svého lékaře. Vždy používejte přípravek ALPROLIX přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem, lékárníkem nebo zdravotní sestrou.
Jestliže jste zapomněl(a) použít přípravek ALPROLIX
Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechanou dávku. Užijte svou dávku co nejdříve si vzpomenete a pak pokračujte v normálním plánu dávkování. Pokud si nejste jistý(á), co máte dělat, zeptejte se svého lékaře, lékárníka nebo nebo zdravotní sestry.
Jestliže jste přestal(a) používat přípravek ALPROLIX
Neukončujte používání přípravku ALPROLIX bez porady se svým lékařem. Pokud ukončíte používání přípravku ALPROLIX, nemusíte být dále chráněn(a) proti krvácení nebo může dojít k tomu, že se stávající krvácení nezastaví.
Máte-li jakékoli další otázky týkající se používání tohoto léčivého přípravku, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se objeví závažné, náhlé alergické reakce (anafylaktická reakce), musí být podávání injekce okamžitě ukončeno. Musíte okamžitě kontaktovat svého lékaře, pokud se u Vás objeví některé z následujících příznaků alergických reakcí: otok obličeje, vyrážka, svědění kůže celého těla, kopřivka, tlak na hrudi, problémy s dýcháním, pálení a píchání v místě vpichu injekce, třesavka, zrudnutí, bolest hlavy, celkový pocit nevolnosti, pocit na zvracení, neklid, zrychlený srdeční tep a nízký krevní tlak.
U tohoto léčivého přípravku se mohou objevit následující nežádoucí účinky.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 pacientů): bolest hlavy, necitlivost nebo brnění úst, bolest v boku s krví v moči (obstrukční uropatie).
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 pacientů): závrať, změna chuti, zápach z úst, pocit únavy, bolest v místě podání injekce, rychlý srdeční tep, krev v moči (hematurie), bolest v boku (ledvinová kolika), nízký krevní tlak a snížená chuť k jídlu.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek ALPROLIX uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Přípravek ALPROLIX může být také uchováván při pokojové teplotě (do 30°C) po jedno nepřetržité období nepřesahující 6 měsíců. Zaznamenejte si prosím na vnější obal datum vyjmutí přípravku
ALPROLIX z chladničky a uložení při pokojové teplotě. Po uchovávání při pokojové teplotě nesmí být přípravek vrácen do chladničky.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na vnějším obalu a štítku injekční lahvičky za "EXP". Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce. Nepoužívejte tento léčivý přípravek, pokud byl uchováván při pokojové teplotě déle než 6 měsíců.
Po přípravě by měl být přípravek ALPROLIX použit okamžitě. Pokud nemůžete použít připravený roztok přípravku ALPROLIX okamžitě, měl(a) byste jej použít během 6 hodin, je-li uchováván při pokojové teplotě. Připravený roztok neuchovávejte v chladničce. Chraňte připravený roztok před přímým slunečním světlem.
Připravený roztok bude čirý až mírně opalescentní a bezbarvý. Nepoužívejte tento přípravek, pokud si všimnete, že je zakalený nebo že obsahuje viditelné částice.
Přípravek je určený pouze pro jedno použití.
Nepoužitý roztok zlikvidujte vhodným způsobem. Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek ALPROLIX obsahuje
Prášek:
• Léčivou látkou je eftrenonacogum alfa (rekombinantní koagulační faktor IX, Fc fuzní protein). Jedna injekční lahvička přípravku ALPROLIX obsahuje jmenovité množství eftrenonacogum alfa 250, 500, 1 000, 2 000 nebo 3 000 IU.
• Dalšími složkami jsou sacharóza, histidin, mannitol, polysorbát 20, hydroxid sodný a kyselina chlorovodíková. Pokud jste na dietě s omezeným obsahem sodíku, přečtěte si bod 2.
Rozpouštědlo:
5 ml chlorid sodný a voda na injekci
Jak přípravek ALPROLIX vypadá a co obsahuje toto balení
Přípravek ALPROLIX je dodáván jako prášek a rozpouštědlo pro injekční roztok. Prášek je bílý až téměř bílý prášek nebo koláč (hrudka). Rozpouštědlo poskytované pro přípravu injekčního roztoku je čirý, bezbarvý roztok. Po přípravě je injekční roztok čirý až mírně opalescentní a bezbarvý.
Jedno balení přípravku ALPROLIX obsahuje 1 injekční lahvičku s práškem, 5 ml rozpouštědla v předplněné injekční stříkačce, 1 nástavec pístu, 1 adaptér injekční lahvičky, 1 infuzní soupravu,
2 alkoholové tampóny, 2 náplasti, 1 gázu.
Držitel rozhodnutí o registraci
Biogen Idec Ltd Innovation House 70 Norden Road Maidenhead Berkshire SL64AY Velká Británie
Výrobce
Swedish Orphan Biovitrum AB (publ)
Strandbergsgatan 49 SE-112 76 Stockholm Švédsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci.
Belgie/Belgique/Belgien
Swedish Orphan Biovitrum BVBA
Tél/Tel: + 32 2880 6119 e-mail: benelux@sobi.com
Bt^rapnn
Cyugum Op^aH EuoBmpyM KnoH Etnrapua OOfl Ten.: +420 257 222 034 e-mail: mail.bg@sobi.com
Česká republika
Swedish Orphan Biovitrum s.r.o.
Tel: +420 257 222 034 e-mail: mail.cz@sobi.com
Danmark
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Deutschland
Swedish Orphan Biovitrum GmbH Tel: +49 6103 20269-0 e-mail: mail.de@sobi.com
Eesti
Oy Swedish Orphan Biovitrum Ab c/o CentralPharma Communications OU Tel. +372 6 015 540
e-mail: centralpharma@centralpharma.ee
Lietuva
Oy Swedish Orphan Biovitrum Ab c/o UAB CentralPharma Communications Tel: +370 5 2430444 e-mail: centralpharma@centralpharma.lt
Luxembourg/Luxemburg
Swedish Orphan Biovitrum BVBA
Tél/Tel: + 32 2880 6119 e-mail: benelux@sobi.com
Magyarország
Swedish Orphan Biovitrum s.r.o. Magyarországi Fióktelepe
Tel: +420 257 222 034 e-mail: mail.hu@sobi.com
Malta
Swedish Orphan Biovitrum S.r.l.
Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Nederland
Swedish Orphan Biovitrum BVBA Tel: + 32 2880 6119 e-mail: benelux@sobi.com
Norge
Swedish Orphan Biovitrum AS
Tlf: +47 66 82 34 00 e-mail: mail.no@sobi.com
Eiiáda
Swedish Orphan Biovitrum S.r.l.
Tni: +39 0521 19 111 e-mail: mail.it@sobi.com
Espaňa
Swedish Orphan Biovitrum S.L
Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com
France
Swedish Orphan Biovitrum SARL
Tél: +33 1 85 78 03 40 e-mail: mail.fr@sobi.com
Hrvatska
SWEDISH ORPHAN BIOVITRUM, Glavna Podružnica Zagreb Tel: +420 257 222 034 e-mail: mail.hr@sobi.com
Ireland
Swedish Orphan Biovitrum Ltd
Tel: + 44 1638 722380 e-mail: mail.uk@sobi.com
Island
Swedish Orphan Biovitrum A/S Tlf: + 45 32 96 68 69 e-mail: mail.dk@sobi.com
Italia
Swedish Orphan Biovitrum S.r.l.
Tel: +39 0521 19 111 e-mail: mail.it@sobi.com
Kórcpog
Swedish Orphan Biovitrum S.r.l.
Tni: +39 0521 19 111 e-mail: mail.it@sobi.com
Latvija
Oy Swedish Orphan Biovitrum Ab c/o CentralPharma Communications SIA Tel. +371 67 450 497 e-mail: centralpharma@centralpharma.lv
Osterreich
Swedish Orphan Biovitrum GmbH Tel: +49 6103 20269-0 e-mail: mail.de@sobi.com
Polska
Swedish Orphan Biovitrum Sp. z o.o. Oddzial w Polsce
Tel: +420 257 222 034 e-mail: mail.pl@sobi.com
Portugal
Swedish Orphan Biovitrum S.L
Tel: + 34 913 91 35 80 e-mail: mail.es@sobi.com
Románia
Swedish Orphan Biovitrum s.r.o. Praga - Sucursala Bucuresti
Tel: +420 257 222 034 e-mail: mail.ro@sobi.com
Slovenija
Swedish Orphan Biovitrum s.r.o. - Podružnica v Sloveniji
Tel: +420 257 222 034 e-mail : mail.si@sobi.com
Slovenská republika
Swedish Orphan Biovitrum o.z.
Tel: +420 257 222 034 e-mail: mail.sk@sobi.com
Suomi/Finland
Oy Swedish Orphan Biovitrum Ab Puh/Tel: +358 201 558 840 e-mail: mail.fi@sobi.com
Sverige
Swedish Orphan Biovitrum AB (publ)
Tel: +46 8 697 20 00 e-mail: mail.se@sobi.com
United Kingdom
Swedish Orphan Biovitrum Ltd
Tel: + 44 1638 722380 e-mail: mail.uk@sobi.com
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu. Na těchto stránkách naleznete též odkazy na další webové stránky týkající se vzácných onemocnění a jejich léčby.
Obraťte prosím na stránku příbalové informace s bodem 7. Instrukce pro přípravu a podávání
7. Instrukce pro přípravu a podávání
Postupy uvedené níže popisují návod k přípravě a podávání přípravku ALPROLIX.
Přípravek ALPROLIX se podává v intravenózní (i.v.) (nitrožilní) injekci po rozpuštění prášku na injekci v rozpouštědle dodávaném v předplněné injekční stříkačce. Balení přípravku ALPROLIX obsahuje:
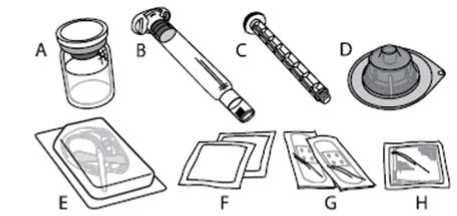
A) 1 injekční lahvička s práškem
B) 5 ml rozpouštědla v předplněné injekční stříkačce
C) 1 nástavec pístu
D) 1 adaptér injekční lahvičky
E) 1 infuzní souprava
F) 2 alkoholové tampóny
G) 2 náplasti
H) 1 gázový polštářek
Přípravek ALPROLIX se nemá míchat s jinými injekčními nebo infuzními roztoky.
Před otevřením balení si umyjte ruce.
Příprava:
1. Zkontrolujte název a sílu balení, abyste se ujistil(a), že obsahuje správný lék. Zkontrolujte dobu použitelnosti na vnějším obalu přípravku ALPROLIX. Nepoužívejte přípravek po uplynutí doby použitelnosti.
2. Pokud byl přípravek ALPROLIX uchováván v chladničce, ponechte přípravek ALPROLIX (A) a injekční stříkačku s rozpouštědlem (B) zahřát na pokojovou teplotu. Nepoužívejte vnější zdroj tepla.
4.
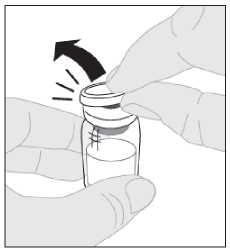
Položte injekční lahvičku na čistý a rovný povrch. Sejměte pojistné plastové víčko z injekční lahvičky s přípravkem
ALPROLIX.
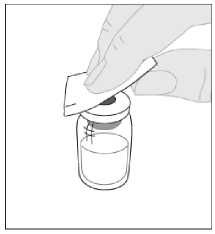
Otřete horní část injekční lahvičky jedním z tampónů napuštěných alkoholem (F), které jsou součástí balení, a nechte ji oschnout. Nedotýkejte se horní části injekční lahvičky a po očištění zabraňte jejímu kontaktu s jakýmkoli předmětem.
5. Sloupněte ochranný papírový kryt z průhledného plastového adaptéru injekční lahvičky (D). Nevyndávejte adaptér z jeho ochranného víčka. Nedotýkejte se vnitřní části balení adaptéru injekční lahvičky.
6. Držte adaptér injekční lahvičky v ochranném víčku a nasaďte jej přímo přes horní část injekční lahvičky. Stlačte pevně adaptér, dokud se nezacvakne na horní část injekční lahvičky a jeho hrot nepronikne přes zátku injekční lahvičky.
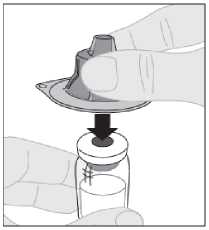
7.
Připojte nástavec pístu (C) na injekční stříkačku s rozpouštědlem zasunutím hrotu pístu do otvoru v pístu injekční stříkačky. Otočte nástavcem pístu pevně ve směru hodinových ručiček, dokud není bezpečně usazen v pístu injekční stříkačky.
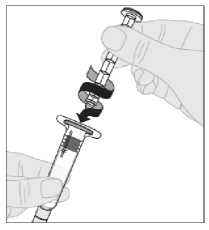
Odlomte bílé bezpečnostní plastové víčko z injekční stříkačky s rozpouštědlem ohnutím v perforaci víčka, dokud se neulomí. Položte víčko stranou horní částí směřující dolů na rovný povrch. Nedotýkejte se vnitřní části víčka ani hrotu injekční stříkačky.
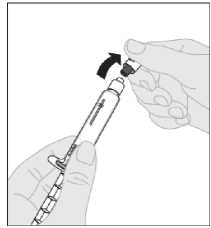
9. Sejměte ochranné víčko z adaptéru a zlikvidujte jej.
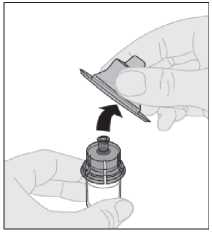
10. Připojte injekční stříkačku s rozpouštědlem na adaptér injekční lahvičky zasunutím hrotu injekční stříkačky do otvoru adaptéru. Pevně zatlačte a otočte injekční stříkačkou ve směru hodinových ručiček, dokud není bezpečně připojena.
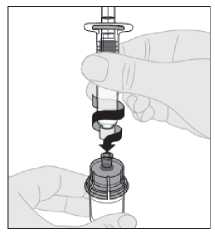
11.
Pomalu stlačte nástavec pístu a vstříkněte veškeré rozpouštědlo do injekční lahvičky s přípravkem ALPROLIX.
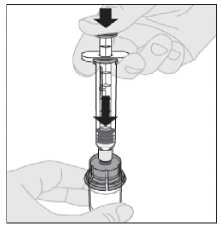
12. S injekční stříkačkou stále připojenou k adaptéru a stlačeným nástavcem pístu rozpusťte prášek jemnými krouživými pohyby injekční lahvičky.
Netřepejte.
13. Před podáním musíte výsledný roztok zkontrolovat zrakem. Roztok by měl být čirý až mírně opalescentní a bezbarvý. Nepoužívejte roztok, pokud je zakalený nebo obsahuje viditelné částice.
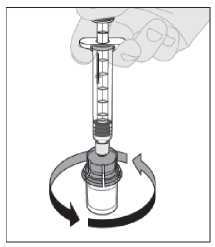
14. S nástavcem pístu injekční lahvičky stále plně stlačeným otočte injekční lahvičku. Pomalu zatáhněte za nástavec pístu, abyste natáhli roztok přes adaptér injekční lahvičky do injekční stříkačky.
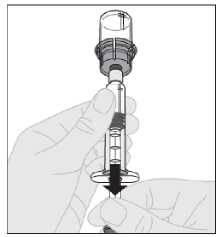
15. Odpojte injekční stříkačku od adaptéru injekční lahvičky jemným tahem a otáčením injekční lahvičky proti směru pohybu hodinových ručiček.
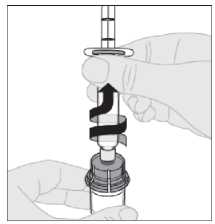
Poznámka: Použijete-li na injekci více než jednu injekční lahvičku s přípravkem ALPROLIX, má být každá injekční lahvička připravena samostatně podle předchozích instrukcí (kroky 1 až 13) a injekční stříkačka s rozpouštědlem má být odstraněna a adaptér injekční lahvičky ponechán na místě. Pro natažení připraveného obsahu každé jednotlivé injekční lahvičky lze použít jednu velkou injekční stříkačku s luerovou koncovkou.
16. Injekční lahvičku a adaptér zlikvidujte.
Poznámka: Pokud nebude roztok použit okamžitě, je třeba pečlivě nasadit zpět víčko injekční stříkačky na hrot injekční stříkačky. Nedotýkejte se hrotu injekční stříkačky ani vnitřní části víčka.
Po přípravě může být přípravek ALPROLIX před podáním uchováván při pokojové teplotě po dobu až 6 hodin. Po uplynutí této doby má být přípravek ALPROLIX zlikvidován. Chraňte před přímým slunečním světlem.
Podávání (intravenózní injekce):
Přípravek ALPROLIX má být podáván pomocí infuzní soupravy (E), která je součástí balení.
|
1. Otevřete balení infuzní soupravy a sejměte víčko na konci hadičky. Připevněte injekční stříkačku s připraveným roztokem přípravku ALPROLIX na konec hadičky infuzní soupravy otočením ve směru hodinových ručiček. |
_ | |||
|
2. Je-li třeba, použijte turniket a připravte místo podání injekce tak, že kůži důkladně otřete pomocí dalšího tampónu napuštěného alkoholem, který je součástí balení. | ||||
|
3. Odstraňte veškerý vzduch z hadiček infuzní soupravy pomalým stlačením nástavce pístu, dokud se tekutina nedostane do jehly infuzní soupravy. Neprotlačujte roztok jehlou. Sejměte průhledný plastový ochranný kryt z jehly. | ||||
|
4. Zaveďte jehlu infuzní soupravy do žíly podle instrukcí svého lékaře nebo zdravotní sestry a odstraňte turniket. Pokud dáváte přednost následující možnosti, můžete použít jednu z náplastí (G), které jsou součástí balení, pro přidržení plastových křidélek jehly v místě podání injekce. Připravený přípravek má být podáván intravenózně injekcí během několika minut. Váš lékař může změnit doporučenou rychlost injekce, aby to pro Vás bylo pohodlnější. | ||||
|
5. Po dokončení podávání injekce a vyjmutí jehly byste měl(a) překlopit chránič jehly a nasunout jej na jehlu. |
/ L-*" LA | |||
|
6. Použitou jehlu, veškerý nepoužitý roztok, injekční stříkačku a prázdnou injekční lahvičku bezpečně zlikvidujte do vhodné odpadní nádobky na zdravotnický materiál, protože tyto předměty mohou zranit ostatní, pokud nejsou správně zlikvidovány. Zařízení nepoužívejte opakovaně. | ||||
39