Advate 500 Iu
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 250 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 50 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle _potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a |
chirurgických výkonech | |
|
Stupeň krvácení / Typ |
Požadovaná hladina |
Frekvence dávek (hodiny) / |
|
chirurgického výkonu |
faktoru VIII (% nebo IU/dl) |
délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do |
20 - 40 |
Opakujte injekce |
|
svalů nebo ústní dutiny. |
každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. | |
|
Rozsáhlejší hemartróza, krvácení |
30 - 60 | |
|
do svalů nebo hematom. |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. | |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší |
30 - 60 |
Každých 24 hodin |
|
Včetně extrakce zubu. |
(12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. | |
|
Větší |
80 - 100 | |
|
(před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). | |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné. Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Komplikace léčby související s použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které _je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního _profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních
hlášení
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo |
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VHI se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
-3---
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních
hlášení
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Není známo | ||
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenán na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 5 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 5 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterililní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela
rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
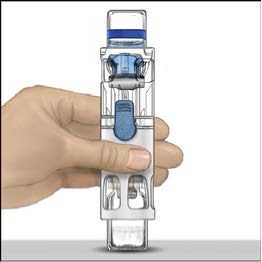
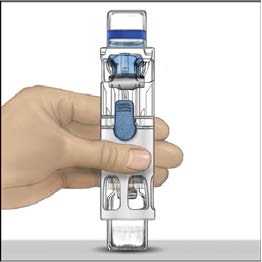
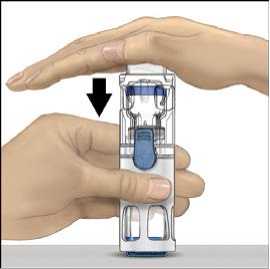
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
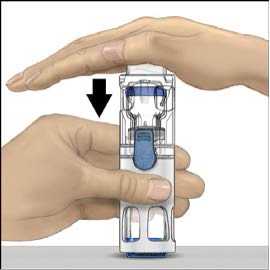
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
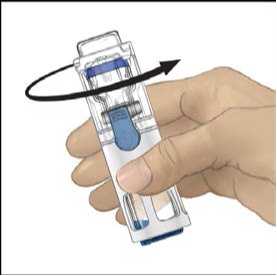
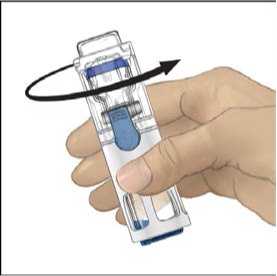
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3
Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/001
EU/1/03/271/011
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
NÁZEV PŘÍPRAVKU
1.
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 100 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle _potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a chirurgických výkonech | ||
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny. |
20 - 40 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom. |
30 - 60 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony Menší Včetně extrakce zubu. Větší |
30 - 60 80 - 100 (před a po operaci) |
Každých 24 hodin (12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné. Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Komplikace léčby související s použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které _je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního _profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních
hlášení
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo |
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VHI se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
-3---
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních
hlášení
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Není známo | ||
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 5 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 5 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela
rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3
Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/002
EU/1/03/271/012
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
1. NÁZEV PŘÍPRAVKU
ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 1000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 200 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a chirurgických výkonech | ||
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny. |
20 - 40 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom. |
30 - 60 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony Menší Včetně extrakce zubu. Větší |
30 - 60 80 - 100 (před a po operaci) |
Každých 24 hodin (12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné. Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Komplikace léčby související s použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního _profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 5 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 5 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT
II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela
rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
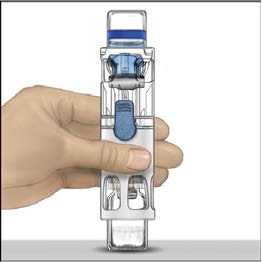
Obr. 1 Obr. 2 Obr. 3
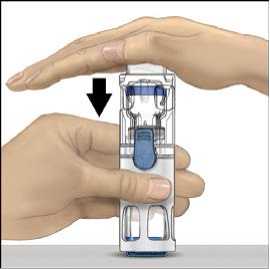
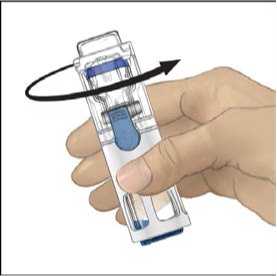
Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/003
EU/1/03/271/013
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
1. NÁZEV PŘÍPRAVKU
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 1500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 300 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a chirurgických výkonech | ||
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny. |
20 - 40 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom. |
30 - 60 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony Menší Včetně extrakce zubu. Větší |
30 - 60 80 - 100 (před a po operaci) |
Každých 24 hodin (12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné. Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Komplikace léčby související s použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního _profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 5 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 5 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela
rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
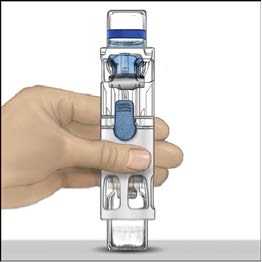
Obr. 1 Obr. 2 Obr. 3
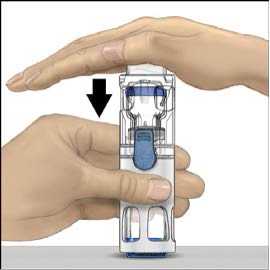
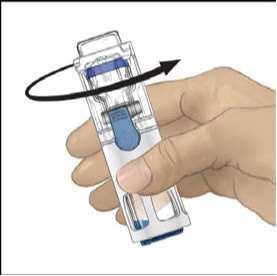
Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/004
EU/1/03/271/014
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
1. NÁZEV PŘÍPRAVKU
ADVATE 2000 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 2000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 400 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a c |
íirurgických výkonech | |
|
Stupeň krvácení / Typ |
Požadovaná hladina |
Frekvence dávek (hodiny) / |
|
chirurgického výkonu |
faktoru VIII (% nebo IU/dl) |
délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do |
20 - 40 |
Opakujte injekce |
|
svalů nebo ústní dutiny. |
každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. | |
|
Rozsáhlejší hemartróza, krvácení |
30 - 60 | |
|
do svalů nebo hematom. |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. | |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší |
30 - 60 |
Každých 24 hodin |
|
Včetně extrakce zubu. |
(12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. | |
|
Větší |
80 - 100 | |
|
(před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). | |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné.
Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická _ populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Komplikace léčby související s použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které _je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního _profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 5 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 5 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT
II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela
rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3
Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/005
EU/1/03/271/015
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
1. NÁZEV PŘÍPRAVKU
ADVATE 3000 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 3000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 600 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a c |
íirurgických výkonech | |
|
Stupeň krvácení / Typ |
Požadovaná hladina |
Frekvence dávek (hodiny) / |
|
chirurgického výkonu |
faktoru VIII (% nebo IU/dl) |
délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do |
20 - 40 |
Opakujte injekce |
|
svalů nebo ústní dutiny. |
každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. | |
|
Rozsáhlejší hemartróza, krvácení |
30 - 60 | |
|
do svalů nebo hematom. |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. | |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší |
30 - 60 |
Každých 24 hodin |
|
Včetně extrakce zubu. |
(12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. | |
|
Větší |
80 - 100 | |
|
(před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). | |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné.
Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická _ populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud
není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Komplikace léčby související s použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které _je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního _profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 5 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 5 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela
rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3
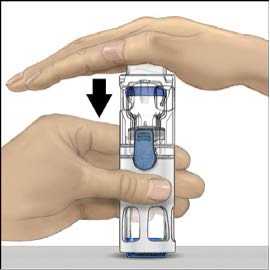
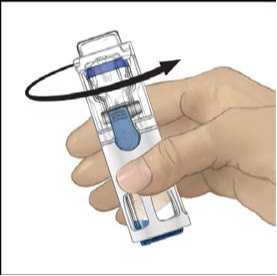
Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/006
EU/1/03/271/016
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
1. NÁZEV PŘÍPRAVKU
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 250 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 125 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a c |
íirurgických výkonech | |
|
Stupeň krvácení / Typ |
Požadovaná hladina |
Frekvence dávek (hodiny) / |
|
chirurgického výkonu |
faktoru VIII (% nebo IU/dl) |
délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do |
20 - 40 |
Opakujte injekce |
|
svalů nebo ústní dutiny. |
každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. | |
|
Rozsáhlejší hemartróza, krvácení |
30 - 60 | |
|
do svalů nebo hematom. |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. | |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší |
30 - 60 |
Každých 24 hodin |
|
Včetně extrakce zubu. |
(12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. | |
|
Větší |
80 - 100 | |
|
(před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). | |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné.
Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická _ populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Použití 2ml provedení nebylo zdokumentováno u pediatrických subjektů mladších 2 let.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Pokud dojde k reakci přecitlivělosti, zbývá kvůli sníženému objemu injikovaného přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci méně času na zareagování, tj. zastavení podávání injekce. Proto je během injikování přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci doporučována opatrnost, a to zejména u dětí.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Nesprávná aplikace přípravku ADVATE
U přípravku ADVATE rekonstituovaného se 2 ml vody na injekci může nesprávná aplikace (intraarteriální nebo paravenózní) vést k lehkým krátkodobým reakcím v místě injekce, jako jsou podlitiny a erytém.
Komplikace léčby související s _použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická _ populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VTTTc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Studie lokální snášenlivosti u králíků ukázala, že přípravek ADVATE rekonstituovaný s 2 ml vody na injekci je po intravenózním podání dobře tolerován. Po intraarteriální aplikaci a paravenózním podání bylo pozorováno přechodné lehké zarudnutí v místě podání. Nebylo však možné zaznamenat žádné související nežádoucí histopatologické změny, což naznačuje přechodný charakter tohoto nálezu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 2 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 2 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela
rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a
Obr. b
Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3

Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/007
EU/1/03/271/017
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
NÁZEV PŘÍPRAVKU
1.
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 250 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle _potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a c |
íirurgických výkonech | |
|
Stupeň krvácení / Typ |
Požadovaná hladina |
Frekvence dávek (hodiny) / |
|
chirurgického výkonu |
faktoru VIII (% nebo IU/dl) |
délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do |
20 - 40 |
Opakujte injekce |
|
svalů nebo ústní dutiny. |
každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. | |
|
Rozsáhlejší hemartróza, krvácení |
30 - 60 | |
|
do svalů nebo hematom. |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. | |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší |
30 - 60 |
Každých 24 hodin |
|
Včetně extrakce zubu. |
(12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. | |
|
Větší |
80 - 100 | |
|
(před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). | |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné. Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Použití 2ml provedení nebylo zdokumentováno u pediatrických subjektů mladších 2 let.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Pokud dojde k reakci přecitlivělosti, zbývá kvůli sníženému objemu injikovaného přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci méně času na zareagování, tj. zastavení podávání injekce. Proto je během injikování přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci doporučována opatrnost, a to zejména u dětí.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Nesprávná aplikace přípravku ADVATE
U přípravku ADVATE rekonstituovaného se 2 ml vody na injekci může nesprávná aplikace (intraarteriální nebo paravenózní) vést k lehkým krátkodobým reakcím v místě injekce, jako jsou podlitiny a erytém.
Komplikace léčby související s _použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které _je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická _ populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Studie lokální snášenlivosti u králíků ukázala, že přípravek ADVATE rekonstituovaný s 2 ml vody na injekci je po intravenózním podání dobře tolerován. Po intraarteriální aplikaci a paravenózním podání bylo pozorováno přechodné lehké zarudnutí v místě podání. Nebylo však možné zaznamenat žádné související nežádoucí histopatologické změny, což naznačuje přechodný charakter tohoto nálezu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 2 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 2 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II)..
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním ADVATE se podává intravenózně po rekonstituci přípravku.
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3

Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/008
EU/1/03/271/018
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
NÁZEV PŘÍPRAVKU
1.
ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 1000 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 500 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle _potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a c |
íirurgických výkonech | |
|
Stupeň krvácení / Typ |
Požadovaná hladina |
Frekvence dávek (hodiny) / |
|
chirurgického výkonu |
faktoru VIII (% nebo IU/dl) |
délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do |
20 - 40 |
Opakujte injekce |
|
svalů nebo ústní dutiny. |
každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. | |
|
Rozsáhlejší hemartróza, krvácení |
30 - 60 | |
|
do svalů nebo hematom. |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. | |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší |
30 - 60 |
Každých 24 hodin |
|
Včetně extrakce zubu. |
(12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. | |
|
Větší |
80 - 100 | |
|
(před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). | |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné. Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Použití 2ml provedení nebylo zdokumentováno u pediatrických subjektů mladších 2 let.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Pokud dojde k reakci přecitlivělosti, zbývá kvůli sníženému objemu injikovaného přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci méně času na zareagování, tj. zastavení podávání injekce. Proto je během injikování přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci doporučována opatrnost, a to zejména u dětí.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Nesprávná aplikace přípravku ADVATE
U přípravku ADVATE rekonstituovaného se 2 ml vody na injekci může nesprávná aplikace (intraarteriální nebo paravenózní) vést k lehkým krátkodobým reakcím v místě injekce, jako jsou podlitiny a erytém.
Komplikace léčby související s _použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které _je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické _pro residua z výrobního _procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická _ populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Studie lokální snášenlivosti u králíků ukázala, že přípravek ADVATE rekonstituovaný s 2 ml vody na injekci je po intravenózním podání dobře tolerován. Po intraarteriální aplikaci a paravenózním podání bylo pozorováno přechodné lehké zarudnutí v místě podání. Nebylo však možné zaznamenat žádné související nežádoucí histopatologické změny, což naznačuje přechodný charakter tohoto nálezu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 2 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 2 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II).
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním ADVATE se podává intravenózně po rekonstituci přípravku
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3

Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/009
EU/1/03/271/019
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
NÁZEV PŘÍPRAVKU
1.
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna lahvička obsahuje 1500 IU lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Po rekonstituci obsahuje přípravek ADVATE přibližně 750 IU/ml lidského koagulačního faktoru VIII (rDNA), octocogum alfa.
Síla (mezinárodní jednotky, IU) se určuje chromogenním testem dle evropského lékopisu. Specifická aktivita přípravku ADVATE je přibližně 4000-10000 IU/mg proteinu.
Octocogum alfa (lidský koagulační faktor VIII (rDNA)) je purifikovaný protein, který má 2332 aminokyselin. Je produkován technologií rekombinace DNA v ovariálních buňkách čínského křečka (CHO). Připravuje se bez přidání jakéhokoli (exogenního) lidského nebo zvířecího proteinu během buněčné kultivace, purifikace nebo konečné formulace.
Pomocné látky se známým účinkem:
0,45 mmol sodíku (10 mg) v lahvičce.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek a rozpouštědlo pro injekční roztok.
Prášek: Bílý až téměř bílý drobivý prášek. Rozpouštědlo: Čirý, bezbarvý roztok.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Léčba a profylaxe krvácení u pacientů s hemofilií A (vrozený nedostatek faktoru VIII). Přípravek ADVATE je indikován pro všechny věkové skupiny.
4.2 Dávkování a způsob podání
Léčba má být zahájena pod dohledem lékaře, který má zkušenosti s léčbou hemofilie. Pro případ anafylaktické reakce musí být ihned k dispozici resuscitační podpora.
Dávkování
Dávka a délka substituční terapie závisí na závažnosti nedostatku faktoru VIII, na místě a rozsahu krvácení a na klinickém stavu pacienta.
Počet jednotek faktoru VIII se vyjadřuje v mezinárodních jednotkách (International Units, IU), které se vztahují ke standardu WHO pro přípravky faktoru VIII. Aktivita faktoru VIII v plazmě se vyjadřuje buď v procentech (ve vztahu k normální lidské plazmě), nebo v IU (podle mezinárodního standardu pro faktor VIII v plazmě).
Jedna mezinárodní jednotka (International Unit - IU) aktivity faktoru VIII se rovná množství faktoru VIII v jednom ml normální lidské plazmy.
Léčba on demand (dle _potřeby)
Výpočet požadované dávky faktoru VIII je založen na empirickém zjištění, že 1 IU faktoru VIII na 1 kg tělesné hmotnosti zvyšuje aktivitu plazmatického faktoru VIII o 2 IU/dl. Požadovaná dávka se určuje podle následujícího vzorce:
Požadované jednotky (IU) = tělesná hmotnost (kg) x požadovaný vzestup faktoru VIII (%) x 0,5.
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka 1 může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech:
|
Tabulka 1 Návod pro dávkování při krvácivých příhodách a c |
íirurgických výkonech | |
|
Stupeň krvácení / Typ |
Požadovaná hladina |
Frekvence dávek (hodiny) / |
|
chirurgického výkonu |
faktoru VIII (% nebo IU/dl) |
délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do |
20 - 40 |
Opakujte injekce |
|
svalů nebo ústní dutiny. |
každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. | |
|
Rozsáhlejší hemartróza, krvácení |
30 - 60 | |
|
do svalů nebo hematom. |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. | |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší |
30 - 60 |
Každých 24 hodin |
|
Včetně extrakce zubu. |
(12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. | |
|
Větší |
80 - 100 | |
|
(před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). | |
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
Jako vodítko k určení dávky a frekvence opakovaných infuzí se doporučuje adekvátní stanovení plazmatických hladin faktoru VIII v průběhu léčby. Zejména v případě větších chirurgických zásahů je přesné sledování substituční terapie pomocí testování plazmatické aktivity faktoru VIII nezbytné. Jednotliví pacienti se mohou lišit svou odpovědí na faktor VIII dosahováním různých hodnot obnovy faktoru VIII in vivo a vykazováním různého poločasu.
Profylaxe
Pro dlouhodobou profylaxi krvácení u pacientů s těžkou hemofilií A jsou obvyklé dávky 20 až 40 IU faktoru VIII na 1 kg tělesné hmotnosti v intervalu dvou až tří dnů.
Pediatrická populace
Při léčbě on demand (dle potřeby) se dávkování u pediatrických pacientů (od narození až do 18 let) neliší od dávkování u dospělých pacientů. U pacientů mladších 6 let se k profylaktické léčbě doporučují dávky faktoru VIII 20 až 50 IU na kg tělesné hmotnosti 3krát až 4krát týdně.
Použití 2ml provedení nebylo zdokumentováno u pediatrických subjektů mladších 2 let.
Způsob podání
ADVATE má být podáván intravenózně. V případě, že aplikaci provádí nezdravotník, je třeba zajistit jeho odpovídající proškolení.
Rychlost podávání by měla být přizpůsobena tomu, jak to pacientovi nejlépe vyhovuje, maximálně 10 ml/min.
Po rekonstituci je roztok čirý, bezbarvý, bez přítomnosti cizích částic a má pH 6,7 až 7,3.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1 nebo na myší nebo křeččí proteiny.
4.4 Zvláštní upozornění a opatření pro použití
Hypersenzitivita
U přípravku ADVATE byly hlášeny reakce přecitlivělosti alergického typu (včetně anafylaxe). Přípravek obsahuje stopy myších a křeččích proteinů. Pokud se symptomy hypersenzitivity objeví, pacienti musí ihned přerušit používání přípravku a kontaktovat svého lékaře. Pacienti mají být informováni o časných příznacích reakcí přecitlivělosti zahrnujících kopřivku, generalizovanou kopřivku, tlak na hrudi, sípání, hypotenzi a anafylaxi,
V případě šoku je nutné postupovat podle standardních léčebných metod pro léčbu šoku.
Pokud dojde k reakci přecitlivělosti, zbývá kvůli sníženému objemu injikovaného přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci méně času na zareagování, tj. zastavení podávání injekce. Proto je během injikování přípravku ADVATE rekonstituovaného ve 2 ml vody na injekci doporučována opatrnost, a to zejména u dětí.
Inhibitory
Tvorba neutralizujících protilátek (inhibitorů) proti faktoru VIII je známou komplikací léčby jedinců s hemofilií A. Tyto inhibitory jsou obvykle IgG imunoglobuliny zaměřené proti prokoagulační aktivitě faktoru VIII, které jsou kvantifikovány v Bethesda jednotkách (Bethesda Units, BU) na ml plazmy s použitím modifikovaného testu. U pacientů, kteří vyvinou inhibitory proti faktoru VIII, se tento stav může projevit nedostatečnou klinickou odpovědí. V těchto případech se doporučuje kontaktovat specializované hemofilické centrum. Riziko vzniku inhibitorů souvisí s rozsahem expozice faktoru VIII, přičemž riziko je nejvyšší během prvních 20 dnů expozice a závisí i na dalších genetických faktorech a faktorech prostředí. Vzácně mohou inhibitory vzniknout po prvních 100 dnech expozice.
Po převedení dříve léčených pacientů s více než 100 dny expozice a vznikem inhibitorů v předchozí anamnéze z jednoho přípravku faktoru VIII na jiný byly zaznamenány případy rekurence inhibitorů (nízkého titru). Proto se po záměně jakéhokoliv přípravku doporučuje u těchto pacientů sledovat, zda se u nich inhibitory znovu neobjeví.
Obecně platí, že všichni pacienti léčení koagulačním faktorem VIII musí být pečlivě sledováni s ohledem na vznik inhibitorů pomocí příslušných klinických pozorování a laboratorních testů. Pokud není dosaženo očekávaných hladin aktivity faktoru VIII v plazmě nebo pokud není krvácení patřičnou dávkou zvládnuto, je třeba provést test na přítomnost inhibitoru faktoru VIII. U pacientů s vysokými hladinami inhibitoru nemusí být substituční terapie faktorem VIII účinná a měly by být zváženy jiné léčebné možnosti. Péče o takové pacienty by měla být vedena lékaři se zkušenostmi v péči o pacienty s hemofilií a inhibitory faktoru VIII.
Nesprávná aplikace přípravku ADVATE
U přípravku ADVATE rekonstituovaného se 2 ml vody na injekci může nesprávná aplikace (intraarteriální nebo paravenózní) vést k lehkým krátkodobým reakcím v místě injekce, jako jsou podlitiny a erytém.
Komplikace léčby související s _použitím katétru
Pokud je potřeba centrální venózní přístupové zařízení (CVAD), je nutné zvážit riziko komplikací ve vztahu k CVAD včetně místních infekcí, bakterémie a trombózy katétru.
Faktory týkající se základu, které _je nutné vzít v úvahu
Po rekonstituci obsahuje tento léčivý přípravek 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu při léčbě pacientů na dietě s kontrolovaným příjmem sodíku.
Důrazně se doporučuje po každém podání přípravku ADVATE pacientovi zaznamenat název a číslo šarže přípravku, aby bylo možné přesně dohledat, jaká šarže léčivého přípravku byla pacientovi podána.
Pediatrická populace:
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce Nebyly provedeny žádné studie interakcí s přípravkem ADVATE.
4.6 Fertilita, těhotenství a kojení
Reprodukční studie u zvířat nebyly s faktorem VIII prováděny. Vzhledem k vzácnému výskytu hemofilie A u žen nejsou zkušenosti s použitím faktoru VIII během těhotenství a kojení. Proto by měl být faktor VIII podáván během těhotenství a kojení pouze v jasných indikacích.
4.7 Účinky na schopnost řídit a obsluhovat stroje
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Klinické studie s přípravkem ADVATE zahrnovaly 418 pacientů s alespoň jednou expozicí přípravku ADVATE; bylo při nich nahlášeno celkem 93 nežádoucích účinků léčiva (NÚ). NÚ, které se vyskytovaly s nejvyšší četností, byly vývoj neutralizujících protilátek faktoru VIII (inhibitorů), bolest hlavy a horečka.
Alergické reakce nebo reakce přecitlivělosti (mezi které patří angioedém, pálení nebo bodání v místě infuze, třesavka, zrudnutí, generalizovaná kopřivka, bolest hlavy, kopřivka, hypotenze, letargie, nevolnost, neklid, tachykardie, tlak na hrudi, brnění, zvracení, sípání) byly pozorovány vzácně a mohou v některých případech progredovat do těžké anafylaxe (včetně šoku).
Může být pozorován vývoj protilátek proti myšímu a/nebo křeččímu proteinu se souvisejícími reakcemi přecitlivělosti.
U pacientů s hemofilií A se mohou vyvinout neutralizující protilátky (inhibitory) proti faktoru VIII. Pokud se takové inhibitory objeví, projeví se tento stav jako nedostatečná klinická odpověď.
V takovém případě se doporučuje kontaktovat specializované hemofilické centrum.
Shrnutí nežádoucích účinků v tabulce
Následující tabulka 2 uvádí četnost výskytu nežádoucích účinků léčiva v klinických studiích a na základě spontánních hlášení. Tabulka je v souladu s klasifikací tříd orgánových systémů MedDRA (TOS a preferované termíny četností).
Kategorie četnosti jsou definovány podle následující konvence: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10000 až < 1/1000) a velmi vzácné (< 1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny v pořadí klesající závažnosti.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Infekce a infestace |
Chřipka |
Méně časté |
|
Méně časté | ||
|
Poruchy krve a lymfatického systému |
Inhibitory faktoru VIIIc |
Časté |
|
Lymfangiitida |
Méně časté | |
|
Poruchy imunitního systému |
Anafylaktická reakce |
Není známo |
|
Přecitlivělost0 |
Není známo | |
|
Poruchy nervového systému |
Bolesti hlavy |
Časté |
|
Závrať |
Méně časté | |
|
Zhoršení paměti |
Méně časté | |
|
Synkopa |
Méně časté | |
|
Méně časté | ||
|
Migréna |
Méně časté | |
|
Dysgeuzie |
Méně časté | |
|
Poruchy oka |
Zánět oka |
Méně časté |
|
Srdeční poruchy |
Palpitace |
Méně časté |
|
Cévní poruchy |
Hematom |
Méně časté |
|
Méně časté | ||
|
Bledost |
Méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
Méně časté | |
|
Gastrointestinální poruchy |
Méně časté | |
|
Bolest v horní části břicha |
Méně časté | |
Vypočtené na základě celkového počtu pacientů, kteří dostávali přípravek ADVATE (418). Neočekávané snížení hladiny koagulačního faktoru VIII se objevilo u jednoho pacienta při kontinuální infuzi ADVATE po chirurgickém výkonu (pooperační dny 10 - 14). Během tohoto období byla po celou dobu zachována hemostáza a hladiny faktoru VIII v plazmě i míra clearance se upravily na dostatečné hodnoty do pooperačního dne 15. Testy na stanovení inhibitoru faktoru VIII, provedené po dokončení kontinuální infuze a při ukončení studie, byly negativní.
|
Tabulka 2 Četnost výskytu nežádoucích účinků v klinických studiích a na základě spontánních hlášení | ||
|
MedDRA standard Třída orgánového systému |
Nežádoucí účinek |
Četnosta |
|
Méně časté | ||
|
Méně časté | ||
|
Poruchy kůže a podkožní tkáně |
Méně časté | |
|
Méně časté | ||
|
Nadměrné pocení |
Méně časté | |
|
Kopřivka |
Méně časté | |
|
Celkové poruchy a reakce v místě aplikace |
Pyrexie |
Časté |
|
Periferní edém |
Méně časté | |
|
Méně časté | ||
|
Hrudní diskomfort |
Méně časté | |
|
Méně časté | ||
|
Nepříjemné pocity |
Méně časté | |
|
Hematom místa punkce cév |
Méně časté | |
|
Únava |
Není známo | |
|
Reakce v místě injekce |
Není známo | |
|
Není známo | ||
|
Vyšetření |
Zvýšený počet monocytů |
Méně časté |
|
Snížení hladiny koagulačního faktoru VIIIb |
Méně časté | |
|
Snížený hematokrit |
Méně časté | |
|
Abnormální laboratorní testy |
Méně časté | |
|
Poranění, otravy a procedurální komplikace |
Procedurální komplikace |
Méně časté |
|
Krvácení po léčebném postupu |
Méně časté | |
|
Reakce v místě podání |
Méně časté | |
a)
b)
c)
Nežádoucí účinky jsou vysvětleny v následující části.
Popis vybraných nežádoucích účinků Vývoj inhibitoru
Vývoj inhibitoru byl hlášen u dříve léčených pacientů (PTP) i u pacientů dříve neléčených (PUP). Podrobnosti naleznete v bodech 5.1 (Farmakologické vlastnosti) a 4.4 (Zvláštní upozornění a opatření pro použití).
Nežádoucí účinky specifické pro residua z výrobního procesu
Z 229 léčených pacientů, u kterých byly zjišťovány protilátky proti CHO (ovariální buňky čínského křečka) buněčnému proteinu, byl u 3 zjištěn statisticky významný vzestupný trend v titrech, u 4 byly nalezeny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí, ale neměl žádné klinické symptomy. Z 229 léčených pacientů, u nichž byly zjišťovány protilátky proti myším IgG, se u 10 vyskytl statisticky významný vzestupný trend, u 2 byly zjištěny ustálené vysoké hladiny nebo přechodné prudké vzestupy a jeden pacient vykazoval obojí. U čtyř z těchto pacientů byly hlášeny izolované případy urtikárie, svědění, vyrážky a lehce zvýšeného počtu eozinofilů během opakovaných expozicí studovanému přípravku.
Hypersenzitivita
Mezi reakce alergického typu patří anafylaxe. Projevují se jako omámenost, parestezie, vyrážka, zrudnutí, otok obličeje, kopřivka a pruritus.
Pediatrická _ populace
V klinických studiích nebyly zjištěny žádné rozdíly specifické pro věk kromě tvorby inhibitorů u dosud neléčených pediatrických pacientů (PUP) a komplikace léčby související s použitím katétru.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Nebyly zaznamenány žádné symptomy předávkování rekombinantním koagulačním faktorem VIII.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihemoragika: krevní koagulační faktory VIII. ATC kód: B02BD02.
Komplex faktoru VIII/von Willebrandova faktoru se skládá ze dvou molekul (faktoru VIII a von Willebrandova faktoru) s různými fyziologickými funkcemi. ADVATE obsahuje rekombinantní koagulační faktor VIII (oktokog alfa), glykoprotein, který je biologicky ekvivalentní glykoproteinu faktoru VIII obsaženém v lidské plazmě.
Oktokog alfa je glykoprotein sestávající z 2332 aminokyselin s přibližnou molekulární hmotností 280 kD. Po infuzi hemofilickému pacientovi se oktokog alfa váže na endogenní von Willebrandův faktor v pacientově krevním oběhu. Aktivovaný faktor VIII působí jako kofaktor aktivovaného faktoru IX, který akceleruje konverzi faktoru X na aktivovaný faktor X. Aktivovaný faktor X konvertuje protrombin na trombin. Trombin potom konvertuje fibrinogen na fibrin a může se vytvořit sraženina. Hemofilie A je na pohlaví závislá dědičná porucha krevní srážlivosti způsobená sníženými hodnotami aktivity faktoru VIII a má za následek silné krvácení do kloubů, svalů nebo vnitřních orgánů, ať už spontánní nebo jako důsledek úrazu nebo chirurgického traumatu. Hladiny faktoru VIII v plazmě se zvyšují substituční terapií, která umožňuje dočasnou úpravu nedostatku faktoru VIII a úpravu sklonu ke krvácení.
Vývoj inhibitoru
Imunogenita přípravku ADVATE byla hodnocena u dříve léčených pacientů. V průběhu klinických studií s přípravkem ADVATE u 233 pediatrických a dospělých pacientů [pediatričtí pacienti (věk 016 let) a dospělí pacienti (věk nad 16 let)] s diagnózou těžké hemofilie A (faktor VIII < 1 %) a s dřívější expozicí koncentrátům faktoru VIII po dobu > 150 dní u dospělých a starších dětí a > 50 dní u dětí mladších 6 let došlo u jednoho pacienta k vytvoření nízkého titru inhibitoru (2,4 BU v modifikovaném Bethesda testu) po 26 dnech expozice přípravku ADVATE. Následné testy inhibitoru u tohoto pacienta po vyřazení ze studie byly negativní. Napříč všemi studiemi byl u dříve léčených pacientů medián expozice přípravku ADVATE 97,0 dnů expozice na pacienta (rozmezí 1 až 709). Celková incidence vývoje jakéhokoliv inhibitoru faktoru VIII (nízká nebo vysoká) byla 0,4 % (1 z 233).
V ukončené nekontrolované studii 060103 se u 16 ze 45 (35,6 %) dříve neléčených pacientů s těžkou hemofilií A (FVIII < 1 %) při alespoň 25 dnech expozice faktoru VIII vyvinuly inhibitory faktoru VIII: u 7 (15,6 %) pacientů se vyvinuly inhibitory o vysokém titru a u 9 (20 %) pacientů se vyvinuly inhibitory o nízkém titru, z nichž 1 byl také klasifikován jako přechodný inhibitor.
Rizikové faktory související s vývojem inhibitoru v této studii zahrnovaly nekavkazskou rasu, inhibitory v rodinné anamnéze a intenzivní léčbu vysokou dávkou v průběhu prvních 20 dnů expozice. U 20 pacientů, kteří neměli žádný z těchto rizikových faktorů, nedošlo k žádnému vývoji inhibitoru.
Nashromáždili jsme data o navození imunotolerance (ITI) u pacientů s inhibitory. V podstudii studie 060103 u dříve neléčených pacientů (PUP) byly léčebné zákroky ITI zdokumentovány u 11 PUP. Retrospektivní analýza tabulky byla provedena u 30 subjektů léčených ITI (studie 060703); sběr registračních dat stále probíhá.
Studie 060201 srovnávala dvě dlouhodobá profylaktická léčebná schémata u 53 dříve léčených pacientů (PTP) - šlo o individualizovaný režim dávkování řízený farmakokinetikou (v rozsahu 20 až 80 UI faktoru VIII na kg hmotnosti v intervalech 72 ± 6 hodin, n = 23) a standardní profylaktický režim dávkování (20 až 40 UI/kg každých 48 ± 6 hodin, n = 30). Cílem režimu dávkování řízeného farmakokinetikou (dle specifického vzorce) bylo dosažení hladin faktoru VIII > 1 % při intervalu mezi dávkami o délce 72 hodin. Údaje z této studie prokazují, že je snížení krvácení u těchto dvou profylaktických režimů dávkování srovnatelné.
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem ADVATE u všech podskupin pediatrické populace s hemofilií A (vrozený deficit faktoru VIII) u „navození imunitní tolerance u pacientů s hemofilií A (vrozený deficit faktoru VIII), u nichž se vyvinuly inhibitory faktoru VIII“ a „léčby a profylaxe krvácení u pacientů s hemofilií A (vrozený deficit faktoru VIII)“ (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Všechny farmakokinetické studie s ADVATE byly prováděny u dříve léčených pacientů s těžkou až středně těžkou hemofilií A (výchozí faktor VIII < 2 %). Vzorky plazmy byly analyzovány v centrální laboratoři s použitím jednostupňového koagulačního testu.
Farmakokinetické parametry, které byly součástí farmakokinetické analytické sady dle protokolu, jsou založeny na údajích od celkem 195 subjektů se závažnou hemofilií A (výchozí hladina faktoru VIII
< 1 %). Farmakokinetické parametry byly shrnuty dle kategorií těchto analýz u kojenců (1 měsíc až
< 2 roky), dětí (2 až < 5 let), starších dětí (5 až < 12 let), adolescentů (12 až < 18 let) a dospělých (18 let nebo více), přičemž věk byl definován jako věk v době infuze ke stanovení farmakokinetiky.
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Celková AUC (IU*h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
|
Úprava přírůstkové recovery v Cmax (IU/dl na IU/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
|
Poločas (h) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
|
Maximální plazmatická koncentrace po infuzi (IU/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
|
Průměrná doba setrvání v oběhu (h) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
|
Tabulka 3 Souhrn farmakokinetických parametrů ADVATE dle věkových skupin se závažnou hemofilií A (výchozí hladina faktoru VIII < 1 %) | |||||
|
Parametr (průměr ± směrodatná odchylka) |
Kojenci (n = 5) |
Děti (n = 30) |
Starší děti (n = 18) |
Adolescenti (n = 33) |
Dospělí (n = 109) |
|
Distribuční objem v ustáleném stavu (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
|
Clearance (ml/kg*h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Vypočteno jako (Cmax - výchozí hladina faktoru VIII) děleno dávkou v IU/kg, kde Cmax je
maximální naměřená hladina faktoru VIII po infuzi.
Bezpečnost a hemostatická účinnost přípravku ADVATE v pediatrické populaci jsou podobné hodnotám v dospělé populaci. Úprava recovery a terminální poločas (ť/2) byly u malých dětí (do 6 let věku) přibližně o 20% nižší než u dospělých, což může být částečně způsobeno vyšším objemem plazmy na kilogram tělesné hmotnosti u mladších pacientů.
Farmakokinetické údaje o ADVATE u dříve neléčených pacientů nejsou v současnosti k dispozici.
5.3 Předklinické údaje vztahující se k bezpečnosti
Neklinické údaje získané na základě farmakologických studií bezpečnosti, akutní toxikologie, toxicity po opakovaném podávání, lokální toxicity a genotoxicity neodhalily žádné zvláštní riziko pro člověka.
Studie lokální snášenlivosti u králíků ukázala, že přípravek ADVATE rekonstituovaný s 2 ml vody na injekci je po intravenózním podání dobře tolerován. Po intraarteriální aplikaci a paravenózním podání bylo pozorováno přechodné lehké zarudnutí v místě podání. Nebylo však možné zaznamenat žádné související nežádoucí histopatologické změny, což naznačuje přechodný charakter tohoto nálezu.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Prášek
Manitol
Chlorid sodný
Histidin
Trehalosa
Chlorid vápenatý
Trometamol
Polysorbát 80
Glutathion (redukovaný)
Rozpouštědlo
Sterilizovaná voda na injekci
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky nebo rozpouštědly.
6.3 Doba použitelnosti
2 roky
Z mikrobiologického hlediska má být přípravek použit okamžitě po rekonstituci. Jeho chemická i fyzikální stabilita před použitím po rekonstituci však byla prokázána při teplotě 25 °C po dobu 3 hodin.
Během doby použitelnosti může být přípravek uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. Konec 6měsíčního uchovávání při pokojové teplotě musí být zaznamenáno na krabičku přípravku. Přípravek nesmí být vrácen zpět do chladničky.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Přípravek ADVATE s pomůckou BAXJECT II: Uchovávejte lahvičku přípravku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek ADVATE v systému BAXJECT III: Uchovávejte těsně uzavřený blistr v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička s práškem i injekční lahvička s 2 ml rozpouštědla jsou ze skla typu I, uzavřené chlorobutylovými pryžovými zátkami. Výrobek se dodává v jedné z následujících konfigurací:
- Přípravek ADVATE s pomůckou BAXJECT II: Každé balení obsahuje injekční lahvičku
s práškem, injekční lahvičku s 2 ml rozpouštědla a pomůcku pro rekonstituci (BAXJECT II)..
- Přípravek ADVATE v systému BAXJECT III: Každé balení obsahuje systém BAXJECT III v těsně uzavřeném blistru připravený k použití (injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla jsou předem sestaveny se systémem pro rekonstituci).
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním ADVATE se podává intravenózně po rekonstituci přípravku
Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice a/nebo nedošlo ke změně jeho barvy.
Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Roztoky, které jsou zakalené nebo obsahují usazeninu, nepoužívejte.
- K podání je zapotřebí použít stříkačku s konektorem luer.
- Použijte do tří hodin po rekonstituci.
- Po rekonstituci přípravek nevracejte do chladničky.
- Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Rekonstituce s pomůckou BAXJECT II
- K rekonstituci použijte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, která je součástí balení.
- Nepoužívejte, pokud pomůcka BAXJECT II, její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
- Má být použit aseptický postup
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT
II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a Obr. b Obr. c

Rekonstituce se systémem BAXJECT III
- Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1 Obr. 2 Obr. 3

Podávání
Použijte aseptický postup
Parenterální léčivé přípravky by se měly před podáním zkontrolovat s ohledem na přítomnost pevných částic, kdykoliv to roztok a obal dovolí. Lze použít pouze čirý a bezbarvý roztok.
1. Sejměte modrý uzávěr z BAXJECT II/BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II/BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu. Před a během podávání ADVATE je třeba měřit tepovou frekvenci. Kdyby došlo k významnému zrychlení, po snížení rychlosti podávání nebo po dočasném přerušení injekce symptomy obvykle rychle zmizí.
(Viz body 4.4 a 4.8).
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Baxter AG Industriestrasse 67 A-1221 Vídeň Rakousko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/010
EU/1/03/271/020
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 2. března 2004
Datum posledního prodloužení registrace: 2. března 2014
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/.
PŘÍLOHA II
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCE ODPOVĚDNÝ ZA
PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky(-ek)
Baxalta Manufacturing Sarl.
Route de Pierre-á-Bot 111 CH-2000 Neuchatel Švýcarsko
Baxalta Manufacturing SARL Singapore Branch
2A Woodlands Industrial Park D Street 2
Singapore 737779
Singapur
Název a adresa výrobců odpovědných za propouštění šarží
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Belgie
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Belgie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2)
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 250 IU oktokogu alfa, přibližně 50 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 250 IU oktokogu alfa, 1 injekční lahvička s 5 ml sterilizované vody na injekci.,1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/001
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 250
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 250 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
5 ml
6. JINÉ
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 250 IU oktokogu alfa, přibližně 50 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 250 IU oktokogu alfa a 1 injekční lahvička s 5 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/011
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 250
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce.
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 250
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ




1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 250
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 500 IU oktokogu alfa, přibližně 100 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 500 IU oktokogu alfa. 1 injekční lahvička s 5 ml sterilizované vody na injekci.,1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/002
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 500
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 500 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
500 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
5 ml
6. JINÉ
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 500 IU oktokogu alfa, přibližně 100 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 500 IU oktokogu alfa a 1 injekční lahvička s 5 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/012
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 500
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 500
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ




1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 500
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 1000 IUprášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1000 IU oktokogu alfa, přibližně 200 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s m1000 IU oktokogu alfa, 1 injekční lahvička s 5 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/003
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
ADVATE 1000 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
1000 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
5 ml
6. JINÉ
ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1000 IU oktokogu alfa, přibližně 200 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 1000 IU oktokogu alfa a 1 injekční lahvička s 5 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/013
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1000
ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 1000
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 500
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1500 IU oktokogu alfa, přibližně 300 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 1500 IU oktokogu alfa, 1 injekční lahvička s 5 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/004
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1500
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
ADVATE 1500 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
1500 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
5 ml
6. JINÉ
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1500 IU oktokogu alfa, přibližně 300 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 1500 IU oktokogu alfa a 1 injekční lahvička s 5 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/014
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1500
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 1500
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 500
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 2000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 2000 IU oktokogu alfa, přibližně 400 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 2000 IU oktokogu alfa, 1 injekční lahvička s 5 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/005
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 2000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
ADVATE 2000 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
2000 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
5 ml
6. JINÉ
ADVATE 2000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 2000 IU oktokogu alfa, přibližně 400 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 2000 IU oktokogu alfa a 1 injekční lahvička s 5 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/015
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 2000
ADVATE 2000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 2000
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
ADVATE 2000

4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 3000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII)
1 injekční lahvička: 3000 IU oktokogu alfa, přibližně 600 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 3000 IU oktokogu alfa, 1 injekční lahvička s 5 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO(A)
EU/1/03/271/006
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 3000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
ADVATE 3000 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
3000 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
5 ml
6. JINÉ
ADVATE 3000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 3000 IU oktokogu alfa, přibližně 600 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 3000 IU oktokogu alfa a 1 injekční lahvička s 5 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/016
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 3000
ADVATE 3000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 5 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 3000
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA(Y) PODÁNÍ
ADVATE 3000

4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET DÁVEK
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 250 IU oktokogu alfa, přibližně 125 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 250 IU oktokogu alfa, 1 injekční lahvička s 2 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/007
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 250
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 250 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
250 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
2 ml
6. JINÉ
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 250 IU oktokogu alfa, přibližně 125 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 250 IU oktokogu alfa a 1 injekční lahvička s 2 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/017
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 250
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 250
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 250
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 500 IU oktokogu alfa, přibližně 250 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 500 IU oktokogu alfa, 1 injekční lahvička s 2 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/008
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 500
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 500 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
500 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
2 ml
6. JINÉ
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 500 IU oktokogu alfa, přibližně 250 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 500 IU oktokogu alfa a 1 injekční lahvička s 2 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/018
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 500
ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 500
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 500
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1000 IU oktokogu alfa, přibližně 500 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 1000 IU oktokogu alfa, 1 injekční lahvička s 2 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/009
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1000
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 1000 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1000 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
2 ml
6. JINÉ
ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1000 IU oktokogu alfa, přibližně 500 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 1000 IU oktokogu alfa a 1 injekční lahvička s 2 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/019
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1000
ADVATE 1000 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 1000
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 1000
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1500 IU oktokogu alfa, přibližně 750 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 1500 IU oktokogu alfa, 1 injekční lahvička s 2 ml sterilizované vody na injekci, 1 pomůcka BAXJECT II.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/010
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1500
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 1500 IU prášek pro injekční roztok. Octocogum alfa IV podání
2. ZPŮSOB PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Pouze k jednorázovému podání.
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
1500 IU oktokogu alfa
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
2 ml
6. JINÉ
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa (rekombinantní lidský koagulační faktor VIII).
1 injekční lahvička: 1500 IU oktokogu alfa, přibližně 750 IU/ml po rekonstituci. Specifická aktivita: přibližně 4000-10000 IU/mg proteinu.
Pomocné látky: manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80, glutathion (redukovaný).
Prášek a rozpouštědlo pro injekční roztok
Obsah: 1 injekční lahvička s 1500 IU oktokogu alfa a 1 injekční lahvička s 2 ml sterilizované vody na injekci předem sestavená se systémem BAXJECT III.
Intravenózní podání, po rekonstituci.
Pouze k jednorázovému podání.
Před použitím si přečtěte příbalovou informaci.
Uchovávejte mimo dohled a dosah dětí.
EXP:
Na konci 6. měsíce, pokud je skladován při pokojové teplotě: Nepoužívejte po uplynutí doby použitelnosti.
Použijte ihned nebo do 3 hodin od rekonstituce.
Uchovávejte v chladničce.
Chraňte před mrazem.
Uchovávejte v původním obalu, aby byl přípravek chráněn před světlem.
Může být uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG A-1221 Vídeň Rakousko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/271/020
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis.
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
ADVATE 1500
ADVATE 1500 IU prášek a rozpouštědlo pro injekční roztok. Octocogum alfa
Baxter AG
EXP:
Č.š.:
Intravenózní podání, po rekonstituci.
Použijte ihned nebo do 3 hodin od rekonstituce,
Pokud je balení otevřeno nebo poškozeno, přípravek nepoužívejte.
Injekční lahvička s práškem a injekční lahvička s 2 ml rozpouštědla předem sestavené se systémem
BAXJECT III.
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADVATE 1500
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Baxter AG
3. POUŽITELNOST
EXP:
4. ČÍSLO ŠARŽE
Č.š.:
5. JINÉ

1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADVATE 1500
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ_
Sterilizovaná voda na injekci
2. ZPŮSOB PODÁNÍ_
3. POUŽITELNOST_
EXP:
4. ČÍSLO ŠARŽE_
Č.š.:
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET_
6 JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: Informace pro uživatele
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 000 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 500 IU prášek a rozpouštědlo pro injekční roztok ADVATE 2 000 IU prášek a rozpouštědlo pro injekční roztok ADVATE 3 000 IU prášek a rozpouštědlo pro injekční roztok
Octocogum alfa (rekombinantní lidský koagulační faktor VIII)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je ADVATE a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete ADVATE používat
3. Jak se ADVATE používá
4. Možné nežádoucí účinky
5. Jak uchovávat ADVATE
6. Obsah balení a další informace
1. CO JE ADVATE A K ČEMU SE POUŽÍVÁ
ADVATE obsahuje léčivou látku oktokog alfa, lidský koagulační faktor VIII produkovaný technologií rekombinace DNA. Faktor VIII je nutný k tvorbě krevní sraženiny a zástavě krvácení. U pacientů s hemofilií A (vrozený nedostatek faktoru VIII) chybí nebo nefunguje správně.
ADVATE se používá k léčbě a prevenci krvácení u pacientů všech věkových skupin s hemofilií A (vrozená porucha krvácení způsobená nedostatkem faktoru VIII).
ADVATE se připravuje bez přidání jakéhokoli lidského nebo zvířecího proteinu během všech fází výrobního procesu.
2. ČEMU MUSÍTE VĚNOVAT POZORNOST, NEŽ ZAČNETE ADVATE POUŽÍVAT
Nepoužívejte ADVATE
- jestliže jste alergický(á) na oktokog alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže jste alergický(á) na myší nebo křeččí proteiny Jestliže si tím nejste jistý(á), zeptejte se svého lékaře.
Upozornění a opatření
Před použitím přípravku ADVATE se poraďte se svým lékařem. Je třeba, abyste informoval(a) svého lékaře, pokud jste byl(a) v minulosti léčen(a) přípravky obsahujícími faktor VIII, zejména pokud se u Vás vyvinuly inhibitory, protože může existovat zvýšené riziko, že se objeví znovu. Inhibitory jsou blokující protilátky proti faktoru VIII, které snižují účinnost ADVATE v prevenci a léčbě krvácení. Vývoj inhibitorů je známou komplikací léčby hemofilie A. Pokud Vaše krvácení nelze zvládnout přípravkem ADVATE, sdělte to ihned svému lékaři.
Existuje vzácné riziko, že vznikne anafylaktická reakce (těžká, náhlá alergická reakce) na ADVATE. Mějte na paměti časné příznaky alergických reakcí, jako je vyrážka, kopřivka, pupeny, celkové svědění, otok rtů a jazyka, ztížené dýchání, sípání, tlak na hrudi, celkový pocit nevolnosti a závratě. Tyto příznaky mohou být časnou známkou anafylaktického šoku, k jehož projevům mohou dále patřit extrémní závratě, ztráta vědomí a extrémní dýchací potíže.
Pokud se některý z těchto příznaků objeví, ihned zastavte injekci a kontaktujte svého lékaře. Těžké příznaky, včetně dýchacích potíží a (téměř) mdlob, vyžadují okamžitou neodkladnou léčbu.
Pacienti s inhibitory faktoru VIII
Jestliže hladina faktoru VIII ve Vaší plazmě při léčbě přípravkem ADVATE nedosáhne očekávané hodnoty nebo není-li krvácení patřičně zvládnuto, může to být způsobeno přítomností inhibitorů faktoru VIII. To bude kontrolovat Váš lékař. Budete možná potřebovat vyšší dávku přípravku ADVATE anebo dokonce jiný přípravek, aby bylo možné krvácení zvládnout. Nezvyšujte celkovou dávku ADVATE abyste dosáhli kontroly krvácení bez předchozí porady se svým lékařem.
Děti a dospívající
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí (od narození do věku 18 let).
Další léčivé přípravky a ADVATE
Informujte svého lékaře o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek ADVATE obsahuje sodík
Tento přípravek obsahuje 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
3. JAK SE ADVATE POUŽÍVÁ
Léčba přípravkem ADVATE bude zahájena lékařem, který má zkušenosti s léčbou pacientů s hemofilií A.
Váš lékař vypočítá dávku přípravku ADVATE (v mezinárodních jednotkách neboli IU) v závislosti na vašem zdravotním stavu a tělesné hmotnosti a podle toho, zda je určen k prevenci nebo léčbě krvácení. Četnost podání bude záviset na tom, jak dobře bude ADVATE ve Vašem případě účinkovat. Léčba přípravkem ADVATE je obvykle doživotní.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Prevence krvácení
Obvyklá dávka oktokogu alfa je 20 až 40 IU na kilogram tělesné hmotnosti, podávaná
každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nezbytné kratší
intervaly mezi dávkami nebo vyšší dávky.
Léčba krvácení
Dávka oktokogu alfa je vypočítána podle Vaší tělesné hmotnosti a požadované hladiny faktoru VIII. Cílová hladina faktoru VIII bude záviset na závažnosti a místě krvácení.
Dávka (IU) = tělesná hmotnost (kg) x žádaný vzestup faktoru VIII (% z normálu) x 0,5
Máte-li pocit, že účinek ADVATE je nedostatečný, sdělte to svému lékaři.
Váš lékař provede určité laboratorní testy, aby se ujistil, že hladina faktoru VIII ve Vaší krvi je dostatečná. To je zvlášť důležité, pokud podstupujete velký chirurgický výkon.
Použití u dětí a dospívajících (od narození do věku 18 let)
Při léčbě krvácení se dávkování u dětí neliší od dospělých pacientů. K prevenci krvácení u dětí mladších 6 let se doporučují dávky 20 až 50 IU na kg tělesné hmotnosti 3- až 4krát týdně. Způsob podávání přípravku ADVATE (intravenózně) se neliší od způsobu podávání tohoto přípravku u dospělých. Může být třeba použít centrální venózní přístupové zařízení (CVAD), které umožní časté infuze produktů faktoru VIII.
Jak se ADVATE podává
ADVATE obvykle podává do žíly (intravenózně) Váš lékař nebo zdravotní sestra. ADVATE můžete aplikovat také Vy nebo jiná vhodná osoba po patřičném vyškolení. Podrobný návod na použití je uveden na konci této příbalové informace.
Jestliže jste použil(a) více přípravku ADVATE, než jste měl(a)
ADVATE používejte vždy přesně podle pokynů Vašeho lékaře. Pokud si nejste jist(á), kontaktujte svého lékaře. Pokud si aplikujete více přípravku ADVATE, než je doporučeno, sdělte to co nejdříve svému lékaři.
Jestliže jste zapomněl(a) použít přípravek ADVATE
Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku. Pokračujte s dalším pravidelným podáním a dále pokračujte podle doporučení Vašeho lékaře.
Jestliže jste přestal(a) užívat přípravek ADVATE
Nepřerušujte používání ADVATE bez konzultace s lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. MOŽNÉ NEŽÁDOUCÍ ÚČINKY
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se objeví závažné, náhlé alergické (anafylaktické) reakce, je nutno okamžitě zastavit injekci. Pokud máte některý z následujících časných příznaků alergických reakcí, musíte okamžitě kontaktovat svého lékaře:
- vyrážka, kopřivka, generalizované svědění
- otok rtů a jazyka
- ztížené dýchání, sípání, tlak na hrudi
- celkový pocit nevolnosti
- závrať a ztráta vědomí
Závažné příznaky, včetně dýchacích obtíží a (téměř) mdlob, vyžadují neodkladnou léčbu.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob)
Inhibitory faktoru VIII, bolest hlavy a horečka.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
Závrať, chřipka, mdloba, abnormální srdeční tep, zarudlé svědící bulky na kůži, nepříjemný pocit na hrudi, modřina v místě injekce, reakce v místě injekce, svědění, zvýšené pocení, neobvyklá pachuť v ústech, návaly horka, migrény, poruchy paměti, zimnice, průjem, nevolnost, zvracení, ztížené dýchání, bolest v krku, infekce lymfatických cév, zbělení kůže, oční zánět, vyrážka, nadměrné pocení, otoky nohou a dolních končetin, snížení počtu červených krvinek, zvýšení určitého typu bílých krvinek (monocytů) a bolest v horní části břicha nebo dolní části hrudníku.
Ve vztahu k chirurgickému výkonu
Katétrové infekce, snížení počtu červených krvinek, otoky končetin a kloubů, prodloužené krvácení po odstranění drénu, snížená hladina faktoru VIII a pooperační zhmožděnina.
Související s centrálním venózním přístupovým zařízením (CVAD)
Katétrové infekce, systémové infekce a lokální srážení krve v místě katétru.
Nežádoucí účinky s neznámou frekvencí (frekvenci nelze z dostupných údajů určit)
Potenciálně život ohrožující reakce (anafylaxe) a jiné alergické reakce (přecitlivělost), celkové poruchy (utahanost, nedostatek energie).
Další nežádoucí účinky u dětí
Mimo vývoje inhibitorů u dříve neléčených dětských pacientů (PUP) a komplikací souvisejících s přítomností katétru nebyly v klinických studiích zaznamenány žádné rozdíly specifické z hlediska věku.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. JAK UCHOVÁVAT ADVATE
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Během doby použitelnosti může být injekční lahvička s práškem uchovávána při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. V takovém případě tento přípravek expiruje na konci tohoto 6měsíčního období nebo po uplynutí data použitelnosti vytištěného na injekční lahvičce výrobku, podle toho, co nastane dříve. Prosím zaznamenejte konec 6měsíčního uchovávání při pokojové teplotě na krabičku přípravku. Po ukončení uchovávání při pokojové teplotě nesmí být přípravek vrácen zpět do chladničky.
Uchovávejte lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Tento přípravek je určen pouze k jednorázovému podání. Všechen nepoužitý roztok vhodným způsobem zlikvidujte.
Přípravek použijte ihned, jakmile se prášek zcela rozpustí.
Po přípravě nevracejte roztok do chladničky.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. OBSAH BALENÍ A DALŠÍ INFORMACE Co ADVATE obsahuje
- Léčivou látkou je octocogum alfa (lidský koagulační faktor VIII, produkovaný technologií rekombinace DNA v buňkách vaječníků čínského křečka). Jedna injekční lahvička s práškem obsahuje nominálně 250, 500, 1 000, 1 500, 2 000 nebo 3 000 IU oktokogu alfa.
- Pomocnými látkami jsou manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80 a glutathion (redukovaný).
Injekční lahvička s rozpouštědlem: 5 ml sterilizované vody na injekci
Jak ADVATE vypadá a obsah balení
ADVATE je bílý až téměř bílý drobivý prášek.
Po rekonstituci je roztok čirý, bezbarvý a neobsahuje cizí částice.
Každé balení také obsahuje pomůcku pro rekonstituci (BAXJECT II).
Držitel rozhodnutí o registraci:
Baxter AG Industriestrasse 67 A-1221 Vídeň, Rakousko
Výrobci:
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Belgie
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00
Lietuva
UAB "Baxter Lithuania''
Tel: +370 5 269 16 90 / +370 5 252 71 00
Bt^rapnn
EaKCTep Etnrapna EOOfl Ten.: +359 2 9808482
Česká republika
Baxter Czech spol.s.r.o. Tel.: +420 225774111
Luxembourg/Luxemburg
Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00
Magyarország
Baxter Hungary Kft.
Tel.: +36 1 202 1980
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OU Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXlába Baxter (Hellas) E.n.E. T^A,: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
Espaňa Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Románia FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Island Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
|
Kónpoq Baxter (Hellas) E.n.E. TnA.: +30 210 28 80 000 |
Sverige Baxalta Sweden AB Tel: +46 8 50 53 26 00 |
|
Latvija SIA BAXTER Latvia Tel.: +371 67 784 784 |
United Kingdom Baxalta UK Limited Tel: +44 1 635 798 777 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema. europa.eu/
Návod na přípravu a podání
Při přípravě a podávání roztoku je třeba dodržovat aseptickou techniku.
Používejte pouze sterilní vodu na injekci a pomůcku pro rekonstituci, které jsou dodávány s každým balením ADVATE. ADVATE nesmí být mísen s dalšími léčivými přípravky nebo rozpouštědly.
Důrazně doporučujeme při každém podání přípravku ADVATE zaznamenat název a číslo šarže přípravku.
Návod na rekonstituci
• Nepoužívejte po uplynutí doby použitelnosti vyznačené na štítcích a krabičce.
• Nepoužívejte, pokud pomůcka BAXJECTII, sterilní bariéra nebo obal vykazují jakékoli
známky poškození, jak znázorňuje symbol
• Po přípravě roztok nevracejte zpět do chladničky.
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT
II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.
Obr. a
Obr. b
Obr. c

Návod na podání
K podání je zapotřebí použít stříkačku s konektorem luer.
Důležitá _ poznámka:
• Nezkoušejte aplikovat injekci sám (sama) pokud jste nebyl(a) řádně proškolen(a) lékařem nebo zdravotní sestrou.
• Připravený roztok před podáním zkontrolujte s ohledem na přítomnost pevných částic a změnu barvy (roztok má být čirý, bezbarvý a bez přítomnosti částic).
Přípravek ADVATE nepoužívejte, není-li roztok zcela čirý nebo není-li zcela rozpuštěn.
1. Sejměte modrý uzávěr z BAXJECT II. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II (Obr. d).
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu (Obr. e).
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte rekonstituovaný roztok intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu (viz bod 4. 6. Možné nežádoucí účinky).
5. Veškerý nespotřebovaný roztok vhodným způsobem zlikvidujte.
Obr. d
Obr. e

Léčba on demand (dle potřeby)
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech.
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny. |
20 - 40 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom. |
30 - 60 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší Včetně extrakce zubu. |
30 - 60 |
Každých 24 hodin (12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. |
|
Větší |
80 - 100 (před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 000 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 500 IU prášek a rozpouštědlo pro injekční roztok ADVATE 2 000 IU prášek a rozpouštědlo pro injekční roztok ADVATE 3 000 IU prášek a rozpouštědlo pro injekční roztok
Octocogum alfa (rekombinantní lidský koagulační faktor VIII)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je ADVATE a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete ADVATE používat
3. Jak se ADVATE používá
4. Možné nežádoucí účinky
5. Jak uchovávat ADVATE
6. Obsah balení a další informace
1. CO JE ADVATE A K ČEMU SE POUŽÍVÁ
ADVATE obsahuje léčivou látku oktokog alfa, lidský koagulační faktor VIII produkovaný technologií rekombinace DNA. Faktor VIII je nutný k tvorbě krevní sraženiny a zástavě krvácení. U pacientů s hemofilií A (vrozený nedostatek faktoru VIII) chybí nebo nefunguje správně.
ADVATE se používá k léčbě a prevenci krvácení u pacientů všech věkových skupin s hemofilií A (vrozená porucha krvácení způsobená nedostatkem faktoru VIII).
ADVATE se připravuje bez přidání jakéhokoli lidského nebo zvířecího proteinu během všech fází výrobního procesu.
2. ČEMU MUSÍTE VĚNOVAT POZORNOST, NEŽ ZAČNETE ADVATE POUŽÍVAT
Nepoužívejte ADVATE
- jestliže jste alergický(á) na oktokog alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže jste alergický(á) na myší nebo křeččí proteiny Jestliže si tím nejste jistý(á), zeptejte se svého lékaře.
Upozornění a opatření
Před použitím přípravku ADVATE se poraďte se svým lékařem. Je třeba, abyste informoval(a) svého lékaře, pokud jste byl(a) v minulosti léčen(a) přípravky obsahujícími faktor VIII, zejména pokud se
u Vás vyvinuly inhibitory, protože může existovat zvýšené riziko, že se objeví znovu. Inhibitory jsou blokující protilátky proti faktoru VIII, které snižují účinnost ADVATE v prevenci a léčbě krvácení. Vývoj inhibitorů je známou komplikací léčby hemofilie A. Pokud Vaše krvácení nelze zvládnout přípravkem ADVATE, sdělte to ihned svému lékaři.
Existuje vzácné riziko, že vznikne anafylaktická reakce (těžká, náhlá alergická reakce) na ADVATE. Mějte na paměti časné příznaky alergických reakcí, jako je vyrážka, kopřivka, pupeny, celkové svědění, otok rtů a jazyka, ztížené dýchání, sípání, tlak na hrudi, celkový pocit nevolnosti a závratě. Tyto příznaky mohou být časnou známkou anafylaktického šoku, k jehož projevům mohou dále patřit extrémní závratě, ztráta vědomí a extrémní dýchací potíže.
Pokud se některý z těchto příznaků objeví, ihned zastavte injekci a kontaktujte svého lékaře. Těžké příznaky, včetně dýchacích potíží a (téměř) mdlob, vyžadují okamžitou neodkladnou léčbu.
Pacienti s inhibitory faktoru VIII
Jestliže hladina faktoru VIII ve Vaší plazmě při léčbě přípravkem ADVATE nedosáhne očekávané hodnoty nebo není-li krvácení patřičně zvládnuto, může to být způsobeno přítomností inhibitorů faktoru VIII. To bude kontrolovat Váš lékař. Budete možná potřebovat vyšší dávku přípravku ADVATE anebo dokonce jiný přípravek, aby bylo možné krvácení zvládnout. Nezvyšujte celkovou dávku ADVATE abyste dosáhli kontroly krvácení bez předchozí porady se svým lékařem.
Děti a dospívající
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí (od narození do věku 18 let).
Další léčivé přípravky a ADVATE
Informujte svého lékaře o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek ADVATE obsahuje sodík
Tento přípravek obsahuje 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
3. JAK SE ADVATE POUŽÍVÁ
Léčba přípravkem ADVATE bude zahájena lékařem, který má zkušenosti s léčbou pacientů s hemofilií A.
Váš lékař vypočítá dávku přípravku ADVATE (v mezinárodních jednotkách neboli IU) v závislosti na vašem zdravotním stavu a tělesné hmotnosti a podle toho, zda je určen k prevenci nebo léčbě krvácení. Četnost podání bude záviset na tom, jak dobře bude ADVATE ve Vašem případě účinkovat. Léčba přípravkem ADVATE je obvykle doživotní.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Prevence krvácení
Obvyklá dávka oktokogu alfa je 20 až 40 IU na kilogram tělesné hmotnosti, podávaná
každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nezbytné kratší
intervaly mezi dávkami nebo vyšší dávky.
Léčba krvácení
Dávka oktokogu alfa je vypočítána podle Vaší tělesné hmotnosti a požadované hladiny faktoru VIII. Cílová hladina faktoru VIII bude záviset na závažnosti a místě krvácení.
Dávka (IU) = tělesná hmotnost (kg) x žádaný vzestup faktoru VIII (% z normálu) x 0,5
Máte-li pocit, že účinek ADVATE je nedostatečný, sdělte to svému lékaři.
Váš lékař provede určité laboratorní testy, aby se ujistil, že hladina faktoru VIII ve Vaší krvi je dostatečná. To je zvlášť důležité, pokud podstupujete velký chirurgický výkon.
Použití u dětí a dospívajících (od narození do věku 18 let)
Při léčbě krvácení se dávkování u dětí neliší od dospělých pacientů. K prevenci krvácení u dětí mladších 6 let se doporučují dávky 20 až 50 IU na kg tělesné hmotnosti 3- až 4krát týdně. Způsob podávání přípravku ADVATE (intravenózně) se neliší od způsobu podávání tohoto přípravku u dospělých. Může být třeba použít centrální venózní přístupové zařízení (CVAD), které umožní časté infuze produktů faktoru VIII.
Jak se ADVATE podává
ADVATE obvykle podává do žíly (intravenózně) Váš lékař nebo zdravotní sestra. ADVATE můžete aplikovat také Vy nebo jiná vhodná osoba po patřičném vyškolení. Podrobný návod na použití je uveden na konci této příbalové informace.
Jestliže jste použil(a) více přípravku ADVATE, než jste měl(a)
ADVATE používejte vždy přesně podle pokynů Vašeho lékaře. Pokud si nejste jist(á), kontaktujte svého lékaře. Pokud si aplikujete více přípravku ADVATE, než je doporučeno, sdělte to co nejdříve svému lékaři.
Jestliže jste zapomněl(a) použít přípravek ADVATE
Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku. Pokračujte s dalším pravidelným podáním a dále pokračujte podle doporučení Vašeho lékaře.
Jestliže jste přestal(a) užívat přípravek ADVATE
Nepřerušujte používání ADVATE bez konzultace s lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. MOŽNÉ NEŽÁDOUCÍ ÚČINKY
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se objeví závažné, náhlé alergické (anafylaktické) reakce, je nutno okamžitě zastavit injekci. Pokud máte některý z následujících časných příznaků alergických reakcí, musíte okamžitě kontaktovat svého lékaře:
- vyrážka, kopřivka, generalizované svědění
- otok rtů a jazyka
- ztížené dýchání, sípání, tlak na hrudi
- celkový pocit nevolnosti
- závrať a ztráta vědomí
Závažné příznaky, včetně dýchacích obtíží a (téměř) mdlob, vyžadují neodkladnou léčbu.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob)
Inhibitory faktoru VIII, bolest hlavy a horečka.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
Závrať, chřipka, mdloba, abnormální srdeční tep, zarudlé svědící bulky na kůži, nepříjemný pocit na hrudi, modřina v místě injekce, reakce v místě injekce, svědění, zvýšené pocení, neobvyklá pachuť v ústech, návaly horka, migrény, poruchy paměti, zimnice, průjem, nevolnost, zvracení, ztížené dýchání, bolest v krku, infekce lymfatických cév, zbělení kůže, oční zánět, vyrážka, nadměrné pocení, otoky nohou a dolních končetin, snížení počtu červených krvinek, zvýšení určitého typu bílých krvinek (monocytů) a bolest v horní části břicha nebo dolní části hrudníku.
Ve vztahu k chirurgickému výkonu
Katétrové infekce, snížení počtu červených krvinek, otoky končetin a kloubů, prodloužené krvácení po odstranění drénu, snížená hladina faktoru VIII a pooperační zhmožděnina.
Související s centrálním venózním přístupovým zařízením (CVAD)
Katétrové infekce, systémové infekce a lokální srážení krve v místě katétru.
Nežádoucí účinky s neznámou frekvencí (frekvenci nelze z dostupných údajů určit)
Potenciálně život ohrožující reakce (anafylaxe) a jiné alergické reakce (přecitlivělost), celkové poruchy (utahanost, nedostatek energie).
Další nežádoucí účinky u dětí
Mimo vývoje inhibitorů u dříve neléčených dětských pacientů (PUP) a komplikací souvisejících s přítomností katétru nebyly v klinických studiích zaznamenány žádné rozdíly specifické z hlediska věku.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. JAK UCHOVÁVAT ADVATE
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Během doby použitelnosti může být blistr s přípravkem uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. V takovém případě tento přípravek expiruje na konci tohoto 6měsíčního období nebo po uplynutí data použitelnosti vytištěného na blistru, podle toho, co nastane dříve. Prosím zaznamenejte konec 6měsíčního uchovávání při pokojové teplotě na krabičku přípravku. Po ukončení uchovávání při pokojové teplotě nesmí být přípravek vrácen zpět do chladničky.
Uchovávejte blistr s přípravkem v krabičce, aby byl přípravek chráněn před světlem.
Tento přípravek je určen pouze k jednorázovému podání. Všechen nepoužitý roztok vhodným způsobem zlikvidujte.
Přípravek použijte ihned, jakmile se prášek zcela rozpustí.
Po přípravě nevracejte roztok do chladničky.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. OBSAH BALENÍ A DALŠÍ INFORMACE
Co ADVATE obsahuje
- Léčivou látkou je octocogum alfa (lidský koagulační faktor VIII, produkovaný technologií rekombinace DNA v buňkách vaječníků čínského křečka). Jedna injekční lahvička s práškem obsahuje nominálně 250, 500, 1000, 1 500, 2 000 nebo 3 000 IU oktokogu alfa.
- Pomocnými látkami jsou manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80 a glutathion (redukovaný).
Injekční lahvička s rozpouštědlem: 5 ml sterilizované vody na injekci
Jak ADVATE vypadá a obsah balení
ADVATE je bílý až téměř bílý drobivý prášek.
Po rekonstituci je roztok čirý, bezbarvý a neobsahuje cizí částice.
Držitel rozhodnutí o registraci:
Baxter AG
Industriestrasse 67
A-1221 Vídeň, Rakousko
Výrobci:
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Belgie
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgie/Belgique/Belgien
Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00
Lietuva
UAB "Baxter Lithuania''
Tel: +370 5 269 16 90 / +370 5 252 71 00
Bt^rapnn
EaKCTep Etnrapna EOOfl Ten.: +359 2 9808482
Česká republika
Baxter Czech spol.s.r.o. Tel.: +420 225774111
Luxembourg/Luxemburg
Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00
Magyarország
Baxter Hungary Kft.
Tel.: +36 1 202 1980
Danmark
Baxalta Denmark A/S Tlf: +45 32 70 12 00
Malta
Baxalta UK Limited Tel.: +44 1 635 798 777
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OU Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXlába Baxter (Hellas) E.n.E. T^A,: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
Espaňa Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Románia FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Island Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
|
Kónpoq Baxter (Hellas) E.n.E. TnA.: +30 210 28 80 000 |
Sverige Baxalta Sweden AB Tel: +46 8 50 53 26 00 |
|
Latvija SIA BAXTER Latvia Tel.: +371 67 784 784 |
United Kingdom Baxalta UK Limited Tel: +44 1 635 798 777 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema. europa.eu/
Návod na přípravu a podání
Přípravek ADVATE nesmí být mísen s dalšími léčivými přípravky nebo rozpouštědly.
Důrazně doporučujeme při každém podání přípravku ADVATE zaznamenat název a číslo šarže
přípravku.
Návod na rekonstituci
• Nepoužívejte po uplynutí doby použitelnosti vyznačené na štítcích a krabičce.
• Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
• Po přípravě roztok nevracejte zpět do chladničky.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1

Obr. 2

Obr. 3

Návod na podání
Při podání je nutné dodržovat aseptické postupy.
K podání je zapotřebí použít stříkačku s konektorem luer.
Důležitá poznámka:
• Nezkoušejte aplikovat injekci sám (sama), pokud jste nebyl(a) řádně proškolen(a) lékařem nebo zdravotní sestrou.
• Připravený roztok před podáním zkontrolujte s ohledem na přítomnost pevných částic a změnu barvy (roztok má být čirý, bezbarvý a bez přítomnosti částic).
Přípravek ADVATE nepoužívejte, není-li roztok zcela čirý nebo není-li zcela rozpuštěn.
1. Sejměte modrý uzávěr z BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte rekonstituovaný roztok intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu (viz bod 4. 6. Možné nežádoucí účinky).
5. Veškerý nespotřebovaný roztok vhodným způsobem zlikvidujte.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Léčba on demand (dle potřeby)
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech.
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny. |
20 - 40 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom. |
30 - 60 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Chirurgické výkony | ||
|
Menší Včetně extrakce zubu. |
30 - 60 |
Každých 24 hodin (12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. |
|
Větší |
80 - 100 (před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
Příbalová informace: Informace pro uživatele
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 000 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 500 IU prášek a rozpouštědlo pro injekční roztok
Octocogum alfa (rekombinantní lidský koagulační faktor VIII)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je ADVATE a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete ADVATE používat
3. Jak se ADVATE používá
4. Možné nežádoucí účinky
5. Jak uchovávat ADVATE
6. Obsah balení a další informace
1. CO JE ADVATE A K ČEMU SE POUŽÍVÁ
ADVATE obsahuje léčivou látku oktokog alfa, lidský koagulační faktor VIII produkovaný technologií rekombinace DNA. Faktor VIII je nutný k tvorbě krevní sraženiny a zástavě krvácení. U pacientů s hemofilií A (vrozený nedostatek faktoru VIII) chybí nebo nefunguje správně.
ADVATE se používá k léčbě a prevenci krvácení u pacientů všech věkových skupin s hemofilií A (vrozená porucha krvácení způsobená nedostatkem faktoru VIII).
ADVATE se připravuje bez přidání jakéhokoli lidského nebo zvířecího proteinu během všech fází výrobního procesu.
2. ČEMU MUSÍTE VĚNOVAT POZORNOST, NEŽ ZAČNETE ADVATE POUŽÍVAT
Nepoužívejte ADVATE
- jestliže jste alergický(á) na oktokog alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže jste alergický(á) na myší nebo křeččí proteiny Jestliže si tím nejste jistý(á), zeptejte se svého lékaře.
Upozornění a opatření
Před použitím přípravku ADVATE se poraďte se svým lékařem. Je třeba, abyste informoval(a) svého lékaře, pokud jste byl(a) v minulosti léčen(a) přípravky obsahujícími faktor VIII, zejména pokud se u Vás vyvinuly inhibitory, protože může existovat zvýšené riziko, že se objeví znovu. Inhibitory jsou blokující protilátky proti faktoru VIII, které snižují účinnost ADVATE v prevenci a léčbě krvácení.
Vývoj inhibitorů je známou komplikací léčby hemofilie A. Pokud Vaše krvácení nelze zvládnout přípravkem ADVATE, sdělte to ihned svému lékaři.
Existuje vzácné riziko, že vznikne anafylaktická reakce (těžká, náhlá alergická reakce) na ADVATE. Mějte na paměti časné příznaky alergických reakcí, jako je vyrážka, kopřivka, pupeny, celkové svědění, otok rtů a jazyka, ztížené dýchání, sípání, tlak na hrudi, celkový pocit nevolnosti a závratě. Tyto příznaky mohou být časnou známkou anafylaktického šoku, k jehož projevům mohou dále patřit extrémní závratě, ztráta vědomí a extrémní dýchací potíže.
Pokud se některý z těchto příznaků objeví, ihned zastavte injekci a kontaktujte svého lékaře. Těžké příznaky, včetně dýchacích potíží a (téměř) mdlob, vyžadují okamžitou neodkladnou léčbu.
Pacienti s inhibitory faktoru VIII
Jestliže hladina faktoru VIII ve Vaší plazmě při léčbě přípravkem ADVATE nedosáhne očekávané hodnoty nebo není-li krvácení patřičně zvládnuto, může to být způsobeno přítomností inhibitorů faktoru VIII. To bude kontrolovat Váš lékař. Budete možná potřebovat vyšší dávku přípravku ADVATE anebo dokonce jiný přípravek, aby bylo možné krvácení zvládnout. Nezvyšujte celkovou dávku ADVATE abyste dosáhli kontroly krvácení bez předchozí porady se svým lékařem.
Děti a dospívající
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí (od narození do věku 18 let).
Další léčivé přípravky a ADVATE
Informujte svého lékaře o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek ADVATE obsahuje sodík
Tento přípravek obsahuje 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Nesprávné podání přípravku ADVATE
Je třeba se vyvarovat nesprávnému podání (injekce do tepny nebo mimo žílu), jelikož se v místě injekce mohou objevit lehké krátkodobé reakce, jako jsou podlitiny a zarudnutí.
3. JAK SE ADVATE POUŽÍVÁ
Léčba přípravkem ADVATE bude zahájena lékařem, který má zkušenosti s léčbou pacientů s hemofilií A.
Váš lékař vypočítá dávku přípravku ADVATE (v mezinárodních jednotkách neboli IU) v závislosti na vašem zdravotním stavu a tělesné hmotnosti a podle toho, zda je určen k prevenci nebo léčbě krvácení. Četnost podání bude záviset na tom, jak dobře bude ADVATE ve Vašem případě účinkovat. Léčba přípravkem ADVATE je obvykle doživotní.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Prevence krvácení
Obvyklá dávka oktokogu alfa je 20 až 40 IU na kilogram tělesné hmotnosti, podávaná
každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nezbytné kratší
intervaly mezi dávkami nebo vyšší dávky.
Léčba krvácení
Dávka oktokogu alfa je vypočítána podle Vaší tělesné hmotnosti a požadované hladiny faktoru VIII. Cílová hladina faktoru VIII bude záviset na závažnosti a místě krvácení.
Dávka (IU) = tělesná hmotnost (kg) x žádaný vzestup faktoru VIII (% z normálu) x 0,5
Máte-li pocit, že účinek ADVATE je nedostatečný, sdělte to svému lékaři.
Váš lékař provede určité laboratorní testy, aby se ujistil, že hladina faktoru VIII ve Vaší krvi je dostatečná. To je zvlášť důležité, pokud podstupujete velký chirurgický výkon.
Použití u dětí a dospívajících (od narození do věku 18 let)
Při léčbě krvácení se dávkování u dětí neliší od dospělých pacientů. K prevenci krvácení u dětí mladších 6 let se doporučují dávky 20 až 50 IU na kg tělesné hmotnosti 3- až 4krát týdně. Způsob podávání přípravku ADVATE (intravenózně) se neliší od způsobu podávání tohoto přípravku u dospělých. Může být třeba použít centrální venózní přístupové zařízení (CVAD), které umožní časté infuze produktů faktoru VIII. Toto provedení je k dispozici s 5 a 2 ml rozpouštědla. Pro 2ml provedení však není k dispozici žádná dokumentace pro děti mladší 2 let.
Kvůli sníženému objemu injikovaného přípravku ADVATE rekonstituovaného ve 2 ml je čas k zareagování na reakci přecitlivělosti během podávání injekce kratší, než je tomu v případě použití většího objemu. Proto je během injikování přípravku ADVATE rekonstituovaného ve 2 ml doporučována opatrnost, a to zejména u dětí.
Jak se ADVATE podává
ADVATE obvykle podává do žíly (intravenózně) Váš lékař nebo zdravotní sestra. ADVATE můžete aplikovat také Vy nebo jiná vhodná osoba po patřičném vyškolení. Podrobný návod na použití je uveden na konci této příbalové informace.
Jestliže jste použil(a) více přípravku ADVATE, než jste měl(a)
ADVATE používejte vždy přesně podle pokynů Vašeho lékaře. Pokud si nejste jist(á), kontaktujte svého lékaře. Pokud si aplikujete více přípravku ADVATE, než je doporučeno, sdělte to co nejdříve svému lékaři.
Jestliže jste zapomněl(a) použít přípravek ADVATE
Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku. Pokračujte s dalším pravidelným podáním a dále pokračujte podle doporučení Vašeho lékaře.
Jestliže jste přestal(a) užívat přípravek ADVATE
Nepřerušujte používání ADVATE bez konzultace s lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. MOŽNÉ NEŽÁDOUCÍ ÚČINKY
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se objeví závažné, náhlé alergické (anafylaktické) reakce, je nutno okamžitě zastavit injekci. Pokud máte některý z následujících časných příznaků alergických reakcí, musíte okamžitě kontaktovat svého lékaře:
- vyrážka, kopřivka, generalizované svědění
- otok rtů a jazyka
- ztížené dýchání, sípání, tlak na hrudi
- celkový pocit nevolnosti
- závrať a ztráta vědomí
Závažné příznaky, včetně dýchacích obtíží a (téměř) mdlob, vyžadují neodkladnou léčbu.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob)
Inhibitory faktoru VIII, bolest hlavy a horečka.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
Závrať, chřipka, mdloba, abnormální srdeční tep, zarudlé svědící bulky na kůži, nepříjemný pocit na hrudi, modřina v místě injekce, reakce v místě injekce, svědění, zvýšené pocení, neobvyklá pachuť v ústech, návaly horka, migrény, poruchy paměti, zimnice, průjem, nevolnost, zvracení, ztížené dýchání, bolest v krku, infekce lymfatických cév, zbělení kůže, oční zánět, vyrážka, nadměrné pocení, otoky nohou a dolních končetin, snížení počtu červených krvinek, zvýšení určitého typu bílých krvinek (monocytů) a bolest v horní části břicha nebo dolní části hrudníku.
Ve vztahu k chirurgickému výkonu
Katétrové infekce, snížení počtu červených krvinek, otoky končetin a kloubů, prodloužené krvácení po odstranění drénu, snížená hladina faktoru VIII a pooperační zhmožděnina.
Související s centrálním venózním přístupovým zařízením (CVAD)
Katétrové infekce, systémové infekce a lokální srážení krve v místě katétru.
Nežádoucí účinky s neznámou frekvencí (frekvenci nelze z dostupných údajů určit)
Potenciálně život ohrožující reakce (anafylaxe) a jiné alergické reakce (přecitlivělost), celkové poruchy (utahanost, nedostatek energie).
Další nežádoucí účinky u dětí
Mimo vývoje inhibitorů u dříve neléčených dětských pacientů (PUP) a komplikací souvisejících s přítomností katétru nebyly v klinických studiích zaznamenány žádné rozdíly specifické z hlediska věku.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. JAK UCHOVÁVAT ADVATE
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Během doby použitelnosti může být injekční lahvička s práškem uchovávána při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. V takovém případě tento přípravek expiruje na konci tohoto 6měsíčního období nebo po uplynutí data použitelnosti vytištěného na injekční lahvičce výrobku, podle toho, co nastane dříve. Prosím zaznamenejte konec 6měsíčního uchovávání při pokojové teplotě na krabičku přípravku. Po ukončení uchovávání při pokojové teplotě nesmí být přípravek vrácen zpět do chladničky.
Uchovávejte lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Tento přípravek je určen pouze k jednorázovému podání. Všechen nepoužitý roztok vhodným způsobem zlikvidujte.
Přípravek použijte ihned, jakmile se prášek zcela rozpustí.
Po přípravě nevracejte roztok do chladničky.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. OBSAH BALENÍ A DALŠÍ INFORMACE Co ADVATE obsahuje
- Léčivou látkou je octocogum alfa (lidský koagulační faktor VIII, produkovaný technologií rekombinace DNA v buňkách vaječníků čínského křečka). Jedna injekční lahvička s práškem obsahuje nominálně 250, 500, 1 000 nebo 1 500 IU oktokogu alfa.
- Pomocnými látkami jsou manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80 a glutathion (redukovaný).
Injekční lahvička s rozpouštědlem: 2 ml sterilizované vody na injekci
Jak ADVATE vypadá a obsah balení
ADVATE je bílý až téměř bílý drobivý prášek.
Po rekonstituci je roztok čirý, bezbarvý a neobsahuje cizí částice.
Každé balení také obsahuje pomůcku pro rekonstituci (BAXJECT II).
Držitel rozhodnutí o registraci:
Baxter AG Industriestrasse 67 A-1221 Vídeň, Rakousko
Výrobci:
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Belgie
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Lietuva UAB "Baxter Lithuania'' Tel: +370 5 269 16 90 / +370 5 252 71 00 |
|
Bt^rapnn EaKCTep Etnrapna EOOfl Ten.: +359 2 9808482 |
Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
|
Česká republika Baxter Czech spol.s.r.o. Tel.: +420 225774111 |
Magyarország Baxter Hungary Kft. Tel.: +36 1 202 1980 |
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OU Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXlába Baxter (Hellas) E.n.E. TpL: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
Espaňa Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Románia FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Island Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
Návod na přípravu a podání
Při přípravě a podávání roztoku je třeba dodržovat aseptickou techniku.
Používejte pouze sterilizovanou vodu na injekci a pomůcku pro rekonstituci, které jsou dodávány s každým balením ADVATE. ADVATE nesmí být mísen s dalšími léčivými přípravky nebo rozpouštědly.
Důrazně doporučujeme při každém podání přípravku ADVATE zaznamenat název a číslo šarže přípravku.
Návod na rekonstituci
• Nepoužívejte po uplynutí doby použitelnosti vyznačené na štítcích a krabičce.
• Nepoužívejte, pokud pomůcka BAXJECTII, sterilní bariéra nebo obal vykazují jakékoli
známky poškození, jak znázorňuje symbol
• Po přípravě roztok nevracejte zpět do chladničky.
1. Je-li přípravek ještě uchováván v chladničce, vyjměte lahvičky s práškem ADVATE
a rozpouštědlem z chladničky a nechte je zahřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Pečlivě si umyjte ruce s použitím mýdla a teplé vody.
3. Odstraňte víčka z injekčních lahviček s práškem a rozpouštědlem.
4. Očistěte zátky lihovými tampóny. Položte lahvičky na rovný čistý povrch.
5. Otevřete obal s pomůckou BAXJECT II tak, že odtrhnete papírový kryt, aniž byste se dotýkali vnitřku (Obr. A). Nevyjímejte pomůcku z obalu. Pomůcku BAXJECT II nepoužívejte, pokud její sterilní bariéra nebo obal vykazují jakékoli známky poškození.
6. Otočte obal a průhledným plastovým hrotem propíchněte zátku rozpouštědla. Uchopte obal za okraj a sejměte ho z BAXJECTu II (Obr. B). Nesnímejte modrý uzávěr z pomůcky BAXJECT
II.
7. K rekonstituci mají být použity výhradně sterilizovaná voda na injekci a pomůcka pro rekonstituci, které jsou součástí balení. S BAXJECTem II připojeným k injekční lahvičce
s rozpouštědlem převraťte celý systém tak, aby injekční lahvička s rozpouštědlem byla nahoře. Bílým plastovým hrotem propíchněte zátku lahvičky s práškem ADVATE. Rozpouštědlo se samo pod tlakem natáhne do injekční lahvičky s práškem ADVATE (Obr. C).
8. Rozpusťte materiál jemným krouživým pohybem. Ujistěte se, že je prášek ADVATE zcela rozpuštěn, jinak neprojde veškerý rekonstituovaný roztok filtrem pomůcky. Přípravek se rozpouští rychle (obvykle za méně než 1 minutu). Po rekonstituci má být roztok čirý, bezbarvý a bez přítomnosti cizích částic.

Návod na podání
K podání je zapotřebí použít stříkačku s konektorem luer.
Důležitá poznámka:
• Nezkoušejte aplikovat injekci sám (sama) pokud jste nebyl(a) řádně proškolen(a) lékařem nebo zdravotní sestrou.
• Připravený roztok před podáním zkontrolujte s ohledem na přítomnost pevných částic a změnu barvy (roztok má být čirý, bezbarvý a bez přítomnosti částic).
Přípravek ADVATE nepoužívejte, není-li roztok zcela čirý nebo není-li zcela rozpuštěn.
1. Sejměte modrý uzávěr z BAXJECT II. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT II (Obr. d).
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu (Obr. e).
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte rekonstituovaný roztok intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu (viz bod 4. 6. Možné nežádoucí účinky).
5. Veškerý nespotřebovaný roztok vhodným způsobem zlikvidujte.
Obr. d Obr. e

Léčba on demand (dle potřeby)
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech.
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny. |
20 - 40 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom. |
30 - 60 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší Včetně extrakce zubu. |
30 - 60 |
Každých 24 hodin (12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. |
|
Větší |
80 - 100 (před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
ADVATE 250 IU prášek a rozpouštědlo pro injekční roztok ADVATE 500 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 000 IU prášek a rozpouštědlo pro injekční roztok ADVATE 1 500 IU prášek a rozpouštědlo pro injekční roztok
Octocogum alfa (rekombinantní lidský koagulační faktor VIII)
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je ADVATE a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete ADVATE používat
3. Jak se ADVATE používá
4. Možné nežádoucí účinky
5. Jak uchovávat ADVATE
6. Obsah balení a další informace
1. CO JE ADVATE A K ČEMU SE POUŽÍVÁ
ADVATE obsahuje léčivou látku oktokog alfa, lidský koagulační faktor VIII produkovaný technologií rekombinace DNA. Faktor VIII je nutný k tvorbě krevní sraženiny a zástavě krvácení. U pacientů s hemofilií A (vrozený nedostatek faktoru VIII) chybí nebo nefunguje správně.
ADVATE se používá k léčbě a prevenci krvácení u pacientů všech věkových skupin s hemofilií A (vrozená porucha krvácení způsobená nedostatkem faktoru VIII).
ADVATE se připravuje bez přidání jakéhokoli lidského nebo zvířecího proteinu během všech fází výrobního procesu.
2. ČEMU MUSÍTE VĚNOVAT POZORNOST, NEŽ ZAČNETE ADVATE POUŽÍVAT
Nepoužívejte ADVATE
- jestliže jste alergický(á) na oktokog alfa nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6)
- jestliže jste alergický(á) na myší nebo křeččí proteiny Jestliže si tím nejste jistý(á), zeptejte se svého lékaře.
Upozornění a opatření
Před použitím přípravku ADVATE se poraďte se svým lékařem. Je třeba, abyste informoval(a) svého lékaře, pokud jste byl(a) v minulosti léčen(a) přípravky obsahujícími faktor VIII, zejména pokud se u Vás vyvinuly inhibitory, protože může existovat zvýšené riziko, že se objeví znovu. Inhibitory jsou blokující protilátky proti faktoru VIII, které snižují účinnost ADVATE v prevenci a léčbě krvácení.
Vývoj inhibitorů je známou komplikací léčby hemofilie A. Pokud Vaše krvácení nelze zvládnout přípravkem ADVATE, sdělte to ihned svému lékaři.
Existuje vzácné riziko, že vznikne anafylaktická reakce (těžká, náhlá alergická reakce) na ADVATE. Mějte na paměti časné příznaky alergických reakcí, jako je vyrážka, kopřivka, pupeny, celkové svědění, otok rtů a jazyka, ztížené dýchání, sípání, tlak na hrudi, celkový pocit nevolnosti a závratě. Tyto příznaky mohou být časnou známkou anafylaktického šoku, k jehož projevům mohou dále patřit extrémní závratě, ztráta vědomí a extrémní dýchací potíže.
Pokud se některý z těchto příznaků objeví, ihned zastavte injekci a kontaktujte svého lékaře. Těžké příznaky, včetně dýchacích potíží a (téměř) mdlob, vyžadují okamžitou neodkladnou léčbu.
Pacienti s inhibitory faktoru VIII
Jestliže hladina faktoru VIII ve Vaší plazmě při léčbě přípravkem ADVATE nedosáhne očekávané hodnoty nebo není-li krvácení patřičně zvládnuto, může to být způsobeno přítomností inhibitorů faktoru VIII. To bude kontrolovat Váš lékař. Budete možná potřebovat vyšší dávku přípravku ADVATE anebo dokonce jiný přípravek, aby bylo možné krvácení zvládnout. Nezvyšujte celkovou dávku ADVATE abyste dosáhli kontroly krvácení bez předchozí porady se svým lékařem.
Děti a dospívající
Uvedená varování a bezpečnostní opatření se týkají dospělých i dětí (od narození do věku 18 let).
Další léčivé přípravky a ADVATE
Informujte svého lékaře o všech lécích, které používáte, které jste v nedávné době používal(a) nebo které možná budete používat.
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Řízení dopravních prostředků a obsluha strojů
ADVATE nemá žádný vliv na schopnost řídit nebo obsluhovat stroje.
Přípravek ADVATE obsahuje sodík
Tento přípravek obsahuje 0,45 mmol sodíku (10 mg) v jedné lahvičce. To je třeba vzít v úvahu u pacientů na dietě s kontrolovaným příjmem sodíku.
Nesprávné podání přípravku ADVATE
Je třeba se vyvarovat nesprávnému podání (injekce do tepny nebo mimo žílu), jelikož se v místě injekce mohou objevit lehké krátkodobé reakce, jako jsou podlitiny a zarudnutí.
3. JAK SE ADVATE POUŽÍVÁ
Léčba přípravkem ADVATE bude zahájena lékařem, který má zkušenosti s léčbou pacientů s hemofilií A.
Váš lékař vypočítá dávku přípravku ADVATE (v mezinárodních jednotkách neboli IU) v závislosti na vašem zdravotním stavu a tělesné hmotnosti a podle toho, zda je určen k prevenci nebo léčbě krvácení. Četnost podání bude záviset na tom, jak dobře bude ADVATE ve Vašem případě účinkovat. Léčba přípravkem ADVATE je obvykle doživotní.
Vždy používejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Prevence krvácení
Obvyklá dávka oktokogu alfa je 20 až 40 IU na kilogram tělesné hmotnosti, podávaná
každé 2 až 3 dny. V některých případech, zejména u mladších pacientů, mohou být nezbytné kratší
intervaly mezi dávkami nebo vyšší dávky.
Léčba krvácení
Dávka oktokogu alfa je vypočítána podle Vaší tělesné hmotnosti a požadované hladiny faktoru VIII. Cílová hladina faktoru VIII bude záviset na závažnosti a místě krvácení.
Dávka (IU) = tělesná hmotnost (kg) x žádaný vzestup faktoru VIII (% z normálu) x 0,5
Máte-li pocit, že účinek ADVATE je nedostatečný, sdělte to svému lékaři.
Váš lékař provede určité laboratorní testy, aby se ujistil, že hladina faktoru VIII ve Vaší krvi je dostatečná. To je zvlášť důležité, pokud podstupujete velký chirurgický výkon.
Použití u dětí a dospívajících (od narození do věku 18 let)
Při léčbě krvácení se dávkování u dětí neliší od dospělých pacientů. K prevenci krvácení u dětí mladších 6 let se doporučují dávky 20 až 50 IU na kg tělesné hmotnosti 3- až 4krát týdně. Způsob podávání přípravku ADVATE (intravenózně) se neliší od způsobu podávání tohoto přípravku u dospělých. Může být třeba použít centrální venózní přístupové zařízení (CVAD), které umožní časté infuze produktů faktoru VIII. Toto provedení je k dispozici s 5 a 2 ml rozpouštědla. Pro 2ml provedení však není k dispozici žádná dokumentace pro děti mladší 2 let.
Kvůli sníženému objemu injikovaného přípravku ADVATE rekonstituovaného ve 2 ml je čas k zareagování na reakci přecitlivělosti během podávání injekce kratší, než je tomu v případě použití většího objemu. Proto je během injikování přípravku ADVATE rekonstituovaného ve 2 ml doporučována opatrnost, a to zejména u dětí.
Jak se ADVATE podává
ADVATE obvykle podává do žíly (intravenózně) Váš lékař nebo zdravotní sestra. ADVATE můžete aplikovat také Vy nebo jiná vhodná osoba po patřičném vyškolení. Podrobný návod na použití je uveden na konci této příbalové informace.
Jestliže jste použil(a) více přípravku ADVATE, než jste měl(a)
ADVATE používejte vždy přesně podle pokynů Vašeho lékaře. Pokud si nejste jist(á), kontaktujte svého lékaře. Pokud si aplikujete více přípravku ADVATE, než je doporučeno, sdělte to co nejdříve svému lékaři.
Jestliže jste zapomněl(a) použít přípravek ADVATE
Nezdvojujte následující dávku, abyste nahradil(a) vynechanou dávku. Pokračujte s dalším pravidelným podáním a dále pokračujte podle doporučení Vašeho lékaře.
Jestliže jste přestal(a) užívat přípravek ADVATE
Nepřerušujte používání ADVATE bez konzultace s lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. MOŽNÉ NEŽÁDOUCÍ ÚČINKY
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Pokud se objeví závažné, náhlé alergické (anafylaktické) reakce, je nutno okamžitě zastavit injekci. Pokud máte některý z následujících časných příznaků alergických reakcí, musíte okamžitě kontaktovat svého lékaře:
- vyrážka, kopřivka, generalizované svědění
- otok rtů a jazyka
- ztížené dýchání, sípání, tlak na hrudi
- celkový pocit nevolnosti
- závrať a ztráta vědomí
Závažné příznaky, včetně dýchacích obtíží a (téměř) mdlob, vyžadují neodkladnou léčbu.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 osob)
Inhibitory faktoru VIII, bolest hlavy a horečka.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 osob)
Závrať, chřipka, mdloba, abnormální srdeční tep, zarudlé svědící bulky na kůži, nepříjemný pocit na hrudi, modřina v místě injekce, reakce v místě injekce, svědění, zvýšené pocení, neobvyklá pachuť v ústech, návaly horka, migrény, poruchy paměti, zimnice, průjem, nevolnost, zvracení, ztížené dýchání, bolest v krku, infekce lymfatických cév, zbělení kůže, oční zánět, vyrážka, nadměrné pocení, otoky nohou a dolních končetin, snížení počtu červených krvinek, zvýšení určitého typu bílých krvinek (monocytů) a bolest v horní části břicha nebo dolní části hrudníku.
Ve vztahu k chirurgickému výkonu
Katétrové infekce, snížení počtu červených krvinek, otoky končetin a kloubů, prodloužené krvácení po odstranění drénu, snížená hladina faktoru VIII a pooperační zhmožděnina.
Související s centrálním venózním přístupovým zařízením (CVAD)
Katétrové infekce, systémové infekce a lokální srážení krve v místě katétru.
Nežádoucí účinky s neznámou frekvencí (frekvenci nelze z dostupných údajů určit)
Potenciálně život ohrožující reakce (anafylaxe) a jiné alergické reakce (přecitlivělost), celkové poruchy (utahanost, nedostatek energie).
Další nežádoucí účinky u dětí
Mimo vývoje inhibitorů u dříve neléčených dětských pacientů (PUP) a komplikací souvisejících s přítomností katétru nebyly v klinických studiích zaznamenány žádné rozdíly specifické z hlediska věku.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. JAK UCHOVÁVAT ADVATE
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2 °C - 8 °C).
Chraňte před mrazem.
Během doby použitelnosti může být blistr s přípravkem uchováván při pokojové teplotě (do 25 °C) po jedno období nepřesahující 6 měsíců. V takovém případě tento přípravek expiruje na konci
tohoto 6měsíčního období nebo po uplynutí data použitelnosti vytištěného na blistru, podle toho, co nastane dříve. Prosím zaznamenejte konec 6měsíčního uchovávání při pokojové teplotě na krabičku přípravku. Po ukončení uchovávání při pokojové teplotě nesmí být přípravek vrácen zpět do chladničky.
Uchovávejte blistr s přípravkem v krabičce, aby byl přípravek chráněn před světlem.
Tento přípravek je určen pouze k jednorázovému podání. Všechen nepoužitý roztok vhodným způsobem zlikvidujte.
Přípravek použijte ihned, jakmile se prášek zcela rozpustí.
Po přípravě nevracejte roztok do chladničky.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo do domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. OBSAH BALENÍ A DALŠÍ INFORMACE Co ADVATE obsahuje
- Léčivou látkou je octocogum alfa (lidský koagulační faktor VIII, produkovaný technologií rekombinace DNA v buňkách vaječníků čínského křečka). Jedna injekční lahvička s práškem obsahuje nominálně 250, 500, 1 000 nebo 1 500 IU oktokogu alfa.
- Pomocnými látkami jsou manitol, chlorid sodný, histidin, trehalosa, chlorid vápenatý, trometamol, polysorbát 80 a glutathion (redukovaný).
Injekční lahvička s rozpouštědlem: 2 ml sterilizované vody na injekci
Jak ADVATE vypadá a obsah balení
ADVATE je bílý až téměř bílý drobivý prášek.
Po rekonstituci je roztok čirý, bezbarvý a neobsahuje cizí částice.
Držitel rozhodnutí o registraci:
Baxter AG Industriestrasse 67 A-1221 Vídeň
Výrobci:
Baxalta Belgium Manufacturing SA Boulevard René Branquart 80 B-7860 Lessines Belgie
Baxter SA
Boulevard René Branquart 80
B-7860 Lessines
Belgie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
Lietuva UAB "Baxter Lithuania'' Tel: +370 5 269 16 90 / +370 5 252 71 00 |
|
Bt^rapnn EaKCTep Etnrapna EOOfl Ten.: +359 2 9808482 |
Luxembourg/Luxemburg Baxalta Belgium SPRL Tél/Tel: +32 2 892 62 00 |
|
Česká republika Baxter Czech spol.s.r.o. Tel.: +420 225774111 |
Magyarország Baxter Hungary Kft. Tel.: +36 1 202 1980 |
|
Danmark Baxalta Denmark A/S Tlf: +45 32 70 12 00 |
Malta Baxalta UK Limited Tel.: +44 1 635 798 777 |
|
Deutschland Baxalta Deutschland GmbH Tel: +49 89 262077-011 |
Nederland Baxalta Netherlands B.V. Tel: +31 30 799 27 77 |
|
Eesti OU Baxter Estonia Tel.: +372 6 515 120 |
Norge Baxalta Norway AS Tlf: +47 22 585 000 |
|
EXlába Baxter (Hellas) E.n.E. TpL: +30 210 28 80 000 |
Osterreich Baxalta Osterreich GmbH Tel.: +43 1 20100-0 |
|
Espaňa Baxalta Spain S.L. Tel: +34 91 790 42 22 |
Polska Baxter Polska Sp. z o.o. Tel.: +48 22 4883 777 |
|
France Baxalta France S.A.S. Tél: +33 1 70 96 06 00 |
Portugal Baxalta Portugal, Unipessoal, Lda. Tel: +351 21 122 03 00 |
|
Hrvatska Baxter d.o.o. Tel: +386 1 420 16 80 |
Románia FARMACEUTICA REMEDIA SA Tel.: +40 21 321 16 40 |
|
Ireland Baxalta UK Limited Tel: +44 1 635 798 777 |
Slovenija Baxter d.o.o. Tel.: +386 1 420 16 80 |
|
Island Lyfjaver ehf. Sími: +354 533 6100 |
Slovenská republika Baxter Slovakia s.r.o. Tel: +421 2 3210 1150 |
|
Italia Baxalta Italy S.r.l. Tel: +39 06 45224 600 |
Suomi/Finland Baxalta Finland Oy Puh/Tel: +358 201478200 |
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu/
Návod na přípravu a podání
ADVATE nesmí být mísen s dalšími léčivými přípravky nebo rozpouštědly.
Důrazně doporučujeme při každém podání přípravku ADVATE zaznamenat název a číslo šarže
přípravku.
Návod na rekonstituci
• Nepoužívejte po uplynutí doby použitelnosti vyznačené na štítcích a krabičce.
• Pokud víčko zcela neutěsňuje blistr, přípravek nepoužívejte.
• Po přípravě roztok nevracejte zpět do chladničky.
1. Pokud je přípravek stále uložen v chladničce, vyjměte těsně uzavřený blistr (obsahuje lahvičky s práškem a rozpouštědlem předem sestavené se systémem pro rekonstituci) z chladničky a nechejte jej ohřát na pokojovou teplotu (mezi 15 °C a 25 °C).
2. Důkladně si umyjte ruce pomocí mýdla a teplé vody.
3. Otevřete balení přípravku ADVATE odloupnutím víčka. Vyjměte systém BAXJECT III z blistru.
4. Položte přípravek ADVATE na rovný povrch s lahvičkou rozpouštědla umístěnou nahoře (obr. 1). Lahvička rozpouštědla má modrý proužek. Nesundávejte modrý kryt, dokud k tomu v dalším kroku nedostanete pokyn.
5. Jednou rukou přidržujte přípravek ADVATE v systému BAXJECT III, druhou rukou pevně tiskněte lahvičku s rozpouštědlem, dokud systém zcela nezkolabuje a rozpouštědlo nezačne proudit do lahvičky přípravku ADVATE (obr. 2). Systém nenaklánějte, dokud není převod dokončen.
6. Ověřte, že došlo k úplnému převodu rozpouštědla. Jemně kružte lahvičkou, dokud se veškerý obsah nerozpustí. Ujistěte se, že se prášek přípravku ADVATE úplně rozpustil, jinak přes filtr zařízení neprojde veškerý rekonstituovaný roztok. Přípravek se rozpustí rychle (obvykle za méně než 1 minutu). Po rekonstituci by měl být roztok čirý, bezbarvý a neměl by obsahovat cizí částice.
Obr. 1

Obr. 2

Obr. 3

Návod na podání
Při podání je nutné dodržovat aseptické postupy.
K podání je zapotřebí použít stříkačku s konektorem luer.
Důležitá poznámka:
• Nezkoušejte aplikovat injekci sám (sama) pokud jste nebyl(a) řádně proškolen(a) lékařem nebo zdravotní sestrou.
• Připravený roztok před podáním zkontrolujte s ohledem na přítomnost pevných částic a změnu barvy (roztok má být čirý, bezbarvý a bez přítomnosti částic).
Přípravek ADVATE nepoužívejte, není-li roztok zcela čirý nebo není-li zcela rozpuštěn.
1. Sejměte modrý uzávěr z BAXJECT III. Nenatahujte vzduch do stříkačky. Stříkačku napojte na BAXJECT III.
2. Obraťte systém (injekční lahvičku s rekonstituovaným roztokem nahoru). Natáhněte rekonstituovaný roztok do stříkačky pomalým tahem pístu.
3. Odpojte stříkačku.
4. Ke stříkačce připojte jehlu s motýlkem. Injikujte rekonstituovaný roztok intravenózně. Roztok má být podáván pomalu, rychlostí, která vyhovuje pacientovi, maximálně 10 ml za minutu (viz bod 4. 6. Možné nežádoucí účinky).
5. Veškerý nespotřebovaný roztok vhodným způsobem zlikvidujte.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Léčba on demand (dle potřeby)
V případě následujících krvácivých příhod by během odpovídajícího období neměla aktivita faktoru VIII poklesnout pod stanovenou hladinu plazmatické aktivity (v % normálu nebo IU/dl). Následující tabulka může být vodítkem pro určení dávkování při krvácivých příhodách a chirurgických výkonech.
Dávka a frekvence podávání by se měla upravit podle klinické odpovědi v jednotlivém případě.
Za jistých okolností (např. přítomnost nízkého titru inhibitoru) mohou být nezbytné dávky vyšší než dávky vypočítané podle vzorce.
|
Stupeň krvácení / Typ chirurgického výkonu |
Požadovaná hladina faktoru VIII (% nebo IU/dl) |
Frekvence dávek (hodiny) / délka trvání léčby (dny) |
|
Krvácení | ||
|
Časná hemartróza, krvácení do svalů nebo ústní dutiny. |
20 - 40 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) nejméně 1 den, dokud se krvácení, které se vyznačuje bolestí, nezastaví nebo se nedosáhne vyléčení. |
|
Rozsáhlejší hemartróza, krvácení do svalů nebo hematom. |
30 - 60 |
Opakujte injekce každých 12 až 24 hodin (8 až 24 hodin u pacientů mladších 6 let) po 3 - 4 dny či více, dokud bolest a akutní nemohoucnost nepřejdou. |
|
Život ohrožující krvácení. |
60 - 100 |
Opakujte injekce každých 8 až 24 hodin (6 až 12 hodin u pacientů mladších 6 let), dokud není nebezpečí zažehnáno. |
|
Chirurgické výkony | ||
|
Menší Včetně extrakce zubu. |
30 - 60 |
Každých 24 hodin (12 až 24 hodin u pacientů mladších 6 let), nejméně jeden den, dokud se nedosáhne vyléčení. |
|
Větší |
80 - 100 (před a po operaci) |
Opakujte injekce každých 8 až 24 hodin (6 až 24 hodin u pacientů mladších 6 let) až do adekvátního zahojení rány, pak pokračujte v terapii nejméně dalších 7 dní, abyste udrželi aktivitu faktoru VIII na 30 % až 60 % (IU/dl). |
262