Adcetris 50 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
ADCETRIS 50 mg prášek pro koncentrát pro infuzní roztok.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje brentuximabum vedotinum 50 mg.
Po rekonstituci (viz bod 6.6) jeden ml obsahuje brentuximabum vedotinum 5 mg.
ADCETRIS je konjugát protilátky a léku skládající se z monoklonální protilátky namířené proti CD30 (rekombinantní chimérický imunoglobulin G1 [IgG1], produkovaný technologií rekombinantní DNA v ovariálních buňkách čínského křečka), která je kovalentně vázaná na antimikrotubulovou látku monomethylauristatin E (MMAE).
Pomocné látky se známým účinkem
Jedna injekční lahvička obsahuje přibližně 13,2 mg sodíku. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok. Bílý až téměř bílý koláč nebo prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
ADCETRIS je indikován k léčbě dospělých pacientů s relabujícím nebo refrakterním CD30+ Hodgkinovým lymfomem (HL):
1. po autologní transplantaci kmenových buněk (ASCT) nebo
2. po nejméně dvou předchozích terapiích v případech, kdy ASCT nebo kombinovaná chemoterapie nepředstavuje léčebnou možnost.
ADECTRIS je indikován k léčbě dospělých pacientů s CD30+ HL při zvýšeném riziku relapsu nebo progrese po ASCT (viz bod 5.1).
ADCETRIS je indikován k léčbě dospělých pacientů s relabujícím nebo refrakterním systémovým anaplastickým velkobuněčným lymfomem (sALCL).
4.2 Dávkování a způsob podání
Brentuximab vedotin má být podáván pod dohledem lékaře se zkušenostmi s podáváním protinádorových léků.
Dávkování
Doporučená dávka je 1,8 mg/kg podávaných intravenózní infuzí trvající 30 minut každé 3 týdny.
Doporučená počáteční dávka pro opakovanou léčbu pacientů s relabujícím či refrakterním HL nebo sALCL, kteří v minulosti reagovali na léčbu přípravkem ADCETRIS, je 1,8 mg/kg podávaných intravenózní infuzí trvající 30 minut každé 3 týdny. Alternativně lze léčbu zahájit poslední tolerovanou dávkou (viz bod 5.1).
Porucha funkce ledvin
Doporučená počáteční dávka u pacientů s těžkou poruchou funkce ledvin je 1,2 mg/kg podávaných intravenózní infuzí trvající 30 minut každé 3 týdny. U pacientů s poruchou funkce ledvin je třeba pečlivě sledovat případné nežádoucí účinky (viz bod 5.2).
Porucha funkce jater
Doporučená počáteční dávka u pacientů s poruchou funkce jater je 1,2 mg/kg podávaných intravenózní infuzí trvající 30 minut každé 3 týdny. U pacientů s poruchou funkce jater je třeba pečlivě sledovat případné nežádoucí účinky (viz bod 5.2).
U pacientů s tělesnou hmotností vyšší než 100 kg se má pro výpočet dávky použít 100 kg (viz bod 6.6).
Před podáním každé dávky této léčby je třeba zkontrolovat kompletní krevní obraz (viz bod 4.4). Pacienty je třeba sledovat během infuze a po ní (viz bod 4.4).
Léčba má pokračovat až do progrese onemocnění nebo nepřijatelné toxicity (viz bod 4.4).
Pacienti s relabujícím nebo refrakterním HL nebo sALCL, u kterých se dosáhne stabilizace onemocnění nebo lepšího výsledku, má být podáno minimálně 8 cyklů a maximálně až 16 cyklů (přibližně 1 rok) (viz bod 5.1).
U pacientů s HL při zvýšeném riziku relapsu nebo progrese po ASCT by léčba přípravkem ADCETRIS měla být zahájena po zotavení z ASCT na základě posouzení klinického stavu. Těmto pacientům má být podáno až 16 cyklů (viz bod 5.1).
Úpravy dávkování
Neutropenie
Pokud se během léčby vyskytne neutropenie, má být řešena odkladem dávky. Příslušná doporučení pro dávkování jsou uvedena v tabulce 1 níže (viz též bod 4.4).
Tabulka 1: Doporučené dávkování při neutropenii
|
Stupeň závažnosti neutropenie (známky a příznaky [zkrácený popis CTCAEa]) |
Úprava dávkovacího schématu |
|
Stupeň 1 (<LLN - 1500/mm3 <LLN - 1,5 x 109/l) nebo Stupeň 2 (<1500 - 1000/mm3 <1,5 - 1,0 x 109/l) |
Pokračovat ve stejné dávce a schématu |
|
Stupeň 3 (<1000 - 500/mm3 <1,0 - 0,5 x 109/l) nebo Stupeň 4 (<500/mm3 <0,5 x 109/l) |
Vynechat dávku až do návratu toxicity na < stupeň 2 nebo do výchozího stavu, poté pokračovat v léčbě ve stejné dávce a schématu b. U pacientů, u kterých se vyskytla neutropenie stupně 3 nebo stupně 4, zvážit v následujících cyklech podpůrnou léčbu růstovými faktory (G-CSF nebo GM-CSF). |
a' Klasifikace podle Obecných terminologických kritérií pro nežádoucí účinky (CTCAE) National
Cancer Institute (NCI) verze 3.0; viz neutrofily/granulocyty; LLN = dolní hranice normy b' Pacienti s výskytem lymfopenie stupně 3 nebo 4 mohou v léčbě pokračovat bez přerušení.
Periferní neuropatie
Pokud během léčby dojde k výskytu nebo ke zhoršení periferní senzorické nebo motorické neuropatie, má se postupovat podle příslušných dávkovacích doporučení uvedených v tabulce 2 níže (viz bod 4.4).
Tabulka 2: Doporučené dávkování při nově vzniklé nebo zhoršené periferní senzorické nebo _motorické neuropatii__
|
Závažnost periferní senzorické nebo motorické neuropatie (známky a příznaky [zkrácený popis CTCAEa]) |
Úprava dávky a schématu |
|
Stupeň 1 (parestezie a/nebo ztráta reflexů, bez ztráty funkce) |
Pokračovat ve stejné dávce a schématu |
|
Stupeň 2 (narušení funkce, avšak bez narušení aktivit každodenního života) nebo Stupeň 3 (narušení aktivit každodenního života) |
Odložit dávku až do návratu toxicity na < stupeň 1 nebo do výchozího stavu, poté v léčbě pokračovat sníženou dávkou 1,2 mg/kg každé 3 týdny |
|
Stupeň 4 (senzorická neuropatie, která je zneschopňující, nebo motorická neuropatie, která je život ohrožující nebo vede k paralýze) |
Přerušit léčbu |
a' Klasifikace podle Obecných terminologických kritérií pro nežádoucí účinky (CTCAE) National Cancer Institute (NCI) verze 3.0; viz neuropatie: motorická; neuropatie: senzorická; a neuropatická bolest.
Starší pacienti
Bezpečnost a účinnost u pacientů ve věku 65 let a starších nebyla stanovena. Nejsou dostupné žádné údaje.
Pediatrická populace
Bezpečnost a účinnost u dětí mladších 18 let nebyla dosud stanovena. Nejsou dostupné žádné údaje.
V neklinických studiích byla pozorována deplece thymu (viz bod 5.3).
Způsob podání
Doporučená dávka přípravku ADCETRIS se podává infuzí v průběhu 30 minut.
Návod k rekonstituci a naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
Brentuximab vedotin se nesmí podávat jako intravenózní bolus. Brentuximab vedotin se má podávat samostatnou intravenózní linkou a nesmí být mísen s jinými léčivými přípravky (viz bod 6.2).
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1. Kombinované použití bleomycinu a brentuximab vedotinu vyvolává plicní toxicitu.
4.4 Zvláštní upozornění a opatření pro použití Progresivní multifokální leukoencefalopatie
U pacientů léčených brentuximab vedotinem může dojít k reaktivaci JC viru (John Cunningham virus, JCV) vedoucí k progresivní multifokální leukoencefalopatii (PML) a úmrtí. PML byla hlášena u pacientů, kteří tuto léčbu dostali po předchozím podání více chemoterapeutických režimů. PML je vzácné demyelinizační onemocnění centrálního nervového systému, které je způsobeno reaktivací latentního JCV, a bývá často fatální.
Pacienty je třeba pečlivě sledovat, zda se u nich neobjeví nové nebo zhoršující se neurologické, kognitivní nebo behaviorální známky nebo příznaky, které by mohly ukazovat na PML. V případě jakéhokoli podezření na PML je třeba podávání brentuximab vedotinu pozastavit. Doporučená vyšetření na PML zahrnují neurologické konzilium, zobrazovací vyšetření mozku magnetickou rezonancí s použitím gadolinia a analýzu mozkomíšního moku na přítomnost DNA JC viru pomocí polymerázové řetězové reakce nebo biopsii mozku s průkazem JCV. Negativní PCR na JC virus nevylučuje PML. Pokud není možné stanovit žádnou alternativní diagnózu, mohou být nezbytná další kontrolní vyšetření a hodnocení. V případě potvrzení diagnózy PML má být podávání brentuximab vedotinu trvale přerušeno.
Lékař má věnovat zvláštní pozornost příznakům ukazujícím na PML, kterých by si pacient nemusel všimnout (např. kognitivní, neurologické nebo psychiatrické příznaky).
Pankreatitida
U pacientů léčených brentuximab vedotinem byla pozorována akutní pankreatitida. Byly hlášeny fatální následky.
Pacienty je nutné pečlivě sledovat s ohledem na novou nebo zhoršující se bolest břicha, která může být projevem akutní pankreatitidy. Vyšetření pacienta má zahrnovat fyzikální vyšetření, laboratorní vyšetření amylázy v séru a lipázy v séru a zobrazení břišní krajiny, například ultrazvukem a jinými vhodnými diagnostickými metodami. Podávání brentuximab vedotinu je nutné pozastavit při jakémkoliv podezření na akutní pankreatitidu. Podávání brentuximab vedotinu je třeba ukončit, pokud se potvrdí diagnóza akutní pankreatitidy.
Pulmonální toxicita.
Případy pulmonální toxicity včetně pneumonitidy, intersticiálního plicního onemocnění a syndromu akutní respirační tísně (ARDS), některé s fatálními následky, byly hlášeny u pacientů užívajících brentuximab vedotin. Přestože nebyla zjištěna příčinná souvislost s brentuximab vedotinem, riziko pulmonální toxicity nelze vyloučit. V případě nových nebo zhoršujících se pulmonálních symptomů (např. kašel nebo dyspnoe) je nutné provést rychlé diagnostické vyšetření a pacienty vhodným způsobem léčit. Zvažte zachování dávkování brentuximab vedotinu během vyhodnocování, dokud nedojde k symptomatickému zlepšení.
Závažné infekce a. oportunní infekce
U pacientů léčených brentuximab vedotinem byly hlášeny závažné infekce, jako např. pneumonie, stafylokoková bakteriémie, sepse/septický šok (včetně fatálních dopadů) a herpes zoster a oportunní infekce, jako např. pneumonie vyvolaná Pneumocystis jiroveci a orální kandidóza. Pacienty je třeba během léčby pečlivě sledovat, zda se u nich nevyskytnou případné vážné a oportunní infekce.
Reakce související s infuzí
Byly hlášeny okamžité i opožděné reakce související s infuzí (IRR), jakož i anafylaktické reakce.
Pacienty je třeba během infuze i po ní pečlivě sledovat. Při výskytu anafylaktické reakce je třeba podávání brentuximab vedotinu okamžitě a trvale přerušit a poskytnout příslušnou léčbu.
V případě výskytu IRR je třeba infuzi přerušit a zahájit příslušnou léčbu. Po odeznění symptomů je v infuzi možno pokračovat pomalejší rychlostí. Pacienti, u kterých se v minulosti vyskytla IRR, mají být před dalšími infuzemi premedikováni. Premedikace může zahrnovat paracetamol, antihistaminikum a kortikosteroid.
IRR jsou častější a závažnější u pacientů s protilátkami proti brentuximab vedotinu (viz bod 4.8). Syndrom nádorového rozpadu
U brentuximab vedotinu byl hlášen syndrom nádorového rozpadu (TLS). Riziko syndromu nádorového rozpadu existuje u pacientů s rychle proliferujícím nádorem a vysokou nádorovou zátěží. Tyto pacienty je třeba pečlivě sledovat a postupovat u nich podle doporučených léčebných postupů. Léčba TLS může zahrnovat agresivní hydrataci, monitorování renálních funkcí, korekci elektrolytových abnormalit, léčbu hyperurikemie a podpůrnou péči.
Periferní neuropatie
Léčba brentuximab vedotinem může způsobit periferní neuropatii, a to jak senzorickou, tak motorickou. Periferní neuropatie vyvolaná brentuximab vedotinem je obvykle následkem kumulativní expozice tomuto léčivému přípravku a ve většině případů je reverzibilní.
V pivotních studiích fáze 2 (SG035-0003 a SG035-0004) činil výskyt již existující periferní neuropatie 24 %. Neuropatie vyvolaná léčbou se vyskytla u 56 % populace. V době posledního hodnocení došlo
u většiny pacientů (83%) ke zlepšení nebo vymizení symptomů periferní neuropatie. U pacientů, kteří uváděli periferní neuropatii, byla u 17 % léčba brentuximab vedotinem ukončena, u 13 % došlo ke snížení dávky a u 21 % pacientů došlo k odložení podání dávky.
Výskyt již existující periferní neuropatie u pacientů s relabujícím či refrakterním HL nebo sALCL, kteří podstoupili opakovanou léčbu brentuximab vedotinem, činil 48 %. Neuropatie vyvolaná léčbou se vyskytla u 69 % populace. U většiny pacientů, kteří podstoupili opakovanou léčbu a u kterých se vyskytla periferní neuropatie vyvolaná léčbou (80 %), bylo v době posledního hodnocení zaznamenáno zlepšení nebo vymizení symptomů periferní neuropatie. U 21 % opakovaně léčených pacientů vedla periferní neuropatie k ukončení léčby a u 34 % pacientů k úpravě dávky.
V populaci fáze 3došlo v době posledního hodnocení u většiny pacientů v rameni s brentuximab vedotinem (85 %) ke zlepšení nebo vymizení symptomů periferní neuropatie. U pacientů, kteří uváděli periferní neuropatii, byla u 23 % léčba brentuximab vedotinem ukončena, u 29 % došlo ke snížení dávky a u 22 % pacientů došlo k odložení podání dávky.
Pacienti mají být sledováni, zda se u nich nevyskytnou příznaky neuropatie, jako např. hypestezie, hyperestezie, parestezie, diskomfort, pocit pálení, neuropatická bolest nebo slabost. U pacientů s výskytem nové nebo zhoršující se periferní neuropatie může být nutné odložit podání brentuximab vedotinu a snížit jeho dávku nebo léčbu vysadit (viz bod 4.2).
Hematologické toxicity
Při podávání brentuximab vedotinu se může vyskytnout anémie 3. nebo 4. stupně, trombocytopenie a protrahovaná (>1 týden) neutropenie 3. nebo 4. stupně. Před podáním každé dávky je třeba zkontrolovat kompletní krevní obraz. V případě výskytu neutropenie 3. nebo 4. stupně viz bod 4.2.
Febrilní neutropenie
Při léčbě brentuximab vedotinem byla hlášena febrilní neutropenie (horečka neznámého původu bez klinicky nebo mikrobiologicky dokumentované infekce s absolutním počtem neutrofilů <1,0 x 109/l, horečkou >38,5 °C; viz CTCAE v3). Před podáním každé dávky této léčby je třeba zkontrolovat kompletní krevní obraz. Pacienty je třeba pečlivě sledovat, zda se u nich nevyskytne horečka, a v případě vzniku febrilní neutropenie u nich postupovat podle doporučených léčebných postupů.
Stevens-Johnsonův syndrom a toxická epidermální nekrolýza
Při podávání brentuximab vedotinu byly hlášeny Stevens-Johnsonův syndrom (SJS) a toxická epidermální nekrolýza (TEN). Byly hlášeny případy s následkem smrti. V případě výskytu SJS nebo TEN je třeba léčbu brentuximab vedotinem přerušit a podat příslušnou léčbu.
Gastrointestinální komplikace
U pacientů léčených brentuximab vedotinem byly hlášeny gastrointestinální (GI) komplikace včetně střevní obstrukce, ileu, enterokolitidy, neutropenické kolitidy, eroze, vředu, perforace a krvácení, některé s fatálními následky. V případě nových nebo zhoršení GI symptomů proveďte rychlé diagnostické vyhodnocení a nasaďte vhodnou léčbu.
Hepatotoxicita
U pacientů užívajících brentuximab vedotin byla hlášena hepatotoxicita v podobě zvýšené hladiny alaninaminotransferázy (ALT) a aspartátaminotransferázy (AST). Vyskytly se také vážné případy jaterní toxicity včetně fatálních následků. Riziko mohou také zvýšit preexistující jaterní onemocnění, komorbidity a souběžné podávání léků. U pacientů léčených brentuximab vedotinem je třeba před zahájením léčby otestovat a pravidelně kontrolovat jaterní funkce. Pacienti s hepatotoxicitou mohou vyžadovat oddálení, změnu dávkování nebo vysazení léčby brentuximab vedotinem.
Hyperglykemie
Hyperglykemie byla během klinických hodnocení hlášena u pacientů se zvýšeným indexem tělesné hmotnosti (BMI) s diabetem mellitus v anamnéze nebo bez něj. Hladiny glukózy v séru mají však být pečlivě sledovány u každého pacienta, u kterého se vyskytla epizoda hyperglykemie. Podle potřeby má být podávána antidiabetická léčba.
Poruchy funkce ledvin a jater
Zkušenosti u pacientů s poruchami funkce ledvin a jater jsou omezené. Dostupná data naznačují, že clearance MMAE by mohla být ovlivněna těžkou poruchou funkce ledvin, poruchou funkce jater a nízkými koncentracemi albuminu v séru (viz bod 5.2).
Obsah sodíku v pomocných látkách
Tento léčivý přípravek obsahuje maximálně 2,1 mmol (či 47 mg) sodíku v jedné dávce. To je nutné vzít v úvahu u pacientů dodržujících dietu s omezeným příjmem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Interakce s léčivými přípravky metabolizovanými cestou CYP3A4 (inhibitory/induktory CYP3A4)
Současné podávání brentuximab vedotinu s ketokonazolem, silným inhibitorem CYP3A4 a P-gp, zvyšovalo expozici antimikrotubulové látce MMAE o přibližně 73 % a neměnilo plazmatickou expozici brentuximab vedotinu. Proto může současné podávání brentuximab vedotinu a silných inhibitorů CYP3A4 a P-gp zvýšit incidenci neutropenie. V případě výskytu neutropenie viz tabulka 1: Doporučené dávkování při neutropenii (viz bod 4.2).
Současné podávání brentuximab vedotinu s rifampicinem, silným induktorem CYP3A4, neměnilo plazmatickou expozici brentuximab vedotinu. I když jsou PK data omezená, zdá se, že současné podávání rifampicinu snížilo koncentrace metabolitů MMAE, které mohly být testovány.
Současné podávání midazolamu, substrátu CYP3A4, s brentuximab vedotinem neměnilo metabolismus midazolamu; proto se neočekává, že by brentuximab vedotin modifikoval expozici lékům, které jsou metabolizovány enzymy CYP3A4.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku.
Ženy ve fertilním věku mají během léčby brentuximab vedotinem a ještě 6 měsíců po ukončení terapie používat dvě metody účinné antikoncepce.
Údaje o podávání brentuximab vedotinu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Brentuximab vedotin lze v těhotenství použít pouze tehdy, pokud přínos pro matku převažuje nad možnými riziky pro plod. Pokud těhotná žena potřebuje léčbu, měla by být srozumitelně poučena o potenciálních rizicích pro plod.
Doporučení pro ženy, jejichž partneři jsou léčeni brentuximab vedotinem, viz bod níže týkající se fertility.
Kojení
Údaje o tom, zda se brentuximab vedotin nebo jeho metabolity vylučují do lidského mateřského mléka, nejsou k dispozici.
Riziko pro kojené novorozence / malé děti nelze vyloučit.
Na základě posouzení potenciálního rizika kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit tuto léčbu.
Fertilita
V neklinických studiích vedla léčba brentuximab vedotinem k testikulární toxicitě a může ovlivnit mužskou fertilitu. U MMAE byly prokázány aneugenní vlastnosti (viz bod 5.3). Proto se mužům léčeným tímto lékem doporučuje nechat si před zahájením léčby zmrazit a uložit vzorky spermatu.
Mužům léčeným tímto lékem se nedoporučuje počít dítě během léčby a po dobu 6 měsíců po poslední dávce.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Brentuximab vedotin může mít malý vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Shrnutí bezpečnostního profilu
Bezpečnostní profil ADCETRISu je založen na dostupných údajích z klinických studií, ze Jmenného pacientského programu (NPP, Named Patient Program) a z dosavadních postmarketingových zkušeností. Frekvence nežádoucích účinků popsaná níže a v tabulce 3 byla stanovena z výsledků klinických studií.
ADCETRIS byl podáván jako monoterapie u 160 pacientů ve dvou studiích fáze 2, a to u pacientů s relabujícím nebo refrakterním HL nebo sALCL. Medián počtu podaných cyklů u pacientů s relabujícím nebo s refrakterním HL byl 9 a u pacientů s relabujícím nebo refrakterním sALCL 7. ADCETRIS byl také podáván jako monoterapie 167 pacientům z 329 v randomizované, placebem kontrolované studii fáze 3, a to pacientům s HL při zvýšeném riziku relapsu nebo progrese po ASCT. Medián počtu podaných cyklů v obou ramenech byl 15.
U pacientů léčených tímto léčivým přípravkem byly velmi časté závažné infekce a oportunní infekce (viz bod 4.4). V populaci studie fáze 2 a fáze 3 byly nejčastěji hlášenými oportunními infekcemi herpes zoster a herpex simplex.
Závažnými nežádoucími účinky léku v populaci pivotní studie fáze 2 a fáze 3 byly: pneumonie, syndrom akutní respirační tísně, bolest hlavy, neutropenie, trombocytopenie, zácpa, průjem, zvracení, nauzea, pyrexie, periferní motorická neuropatie, periferní senzorická neuropatie, hyperglykemie, demyelinizační polyneuropatie, syndrom nádorového rozpadu a Stevens-Johnsonův syndrom.
Nejčastěji pozorovanými nežádoucími účinky (>20 %) v populaci pivotní studie fáze 2 a fáze 3 u pacientů dostávajících tuto léčbu byly: periferní senzorická neuropatie, únava, nauzea, průjem, infekce horních cest dýchacích, neutropenie a kašel. Dále byly nežádoucími účinky také pozorovanými u >20 % populace u studií ve fázi 2 zvracení a pyrexie a ve fázi 3 byla pozorována také periferní motorická neuropatie.
Nežádoucí účinky v populaci studie fáze 2 a fáze 3 vedly k ukončení léčby u 23 % 32 % pacientů v tomto pořadí, kteří dostávali brentuximab vedotin. Vážnými nežádoucími účinky, které vedly k ukončení léčby u dvou nebo více pacientů v populaci buď ve fázi 2, nebo fázi 3, byly periferní senzorická neuropatie, periferní motorická neuropatie, demyelinizační polyneuropatie, opakující se Hodgkinova choroba, zvracení a syndrom akutní respirační tísně. Parestézie také vedla k přerušení léčby u dvou nebo více pacientů v populaci buď ve studiích fáze 2, nebo fáze 3.
Údaje o bezpečnosti u pacientů s relabujícím či refrakterním HL, kteří nepodstoupili autologní transplantaci kmenových buněk a byli léčeni doporučenou dávkou 1,8 mg/kg každé tři týdny, získané ve studiích fáze 1 hodnotících zvyšující se dávky a klinických farmakologických studiích (n = 15 pacientů) a v NPP (n = 26 pacientů) (viz bod 5.1) odpovídaly profilu bezpečnosti v pivotních klinických studiích.
Tabulkový seznam nežádoucích účinků
Nežádoucí účinky přípravku ADCETRIS jsou uvedeny podle tříd orgánových systémů a preferovaných termínů databáze MedDRA (viz tabulka 3). V každé třídě orgánových systémů jsou nežádoucí účinky uvedeny podle následujících kategorií frekvence: Velmi časté (>1/10); časté (>1/100 až <1/10); méně časté (>1/1 000 až <1/100); vzácné (>1/10 000 až <1/1 000); velmi vzácné (<1/10 000); není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinky |
|
Infekce a infestace | |
|
Velmi časté: |
Infekce3, infekce horních cest dýchacích |
|
Časté: |
Sepse/septický šok, herpes zoster, pneumonie, herpex simplex |
|
Méně časté: |
Orální kandidóza, pneumonie vyvolaná Pneumocystis jiroveci, stafylokoková bakteriemie |
|
Není známo: |
Progresivní multifokální leukoencefalopatie |
|
Poruchy krve a lymfatického systému | |
|
Velmi časté: |
Neutropenie |
|
Časté: |
Anémie, trombocytopenie |
|
Není známo: |
Febrilní neutropenie |
|
Poruchy imunitního systému | |
|
Není známo: |
Anafylaktická reakce |
|
Poruchy metabolismu a výživy | |
|
Časté: |
Hyperglykemie |
|
Méně časté: |
Syndrom nádorového rozpadu |
|
Poruchy nervového systému | |
|
Velmi časté: |
Periferní senzorická neuropatie, periferní motorická neuropatie |
|
Časté: |
Závratě, demyelinizační polyneuropatie |
|
Respirační, hrudní a mediastinální poruchy | |
|
Velmi časté: | |
|
Gastrointestinální poruchy | |
|
Velmi časté: |
Průjem, nauzea, zvracení, zácpa, bolest břicha |
|
Méně časté: |
Akutní pankreatitida |
|
Poruchy jater a žlučových cest | |
|
Časté |
Zvýšení hladiny alaninaminotransferázy/ aspartátaminotransferázy (ALT/AST) |
|
Poruchy kůže a podkožní tkáně | |
|
Velmi časté: |
Alopecie, pruritus |
|
Časté: | |
|
Vzácné: |
Stevens-Johnsonův syndrom/toxická epidermální nekrolýza |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně | |
|
Velmi časté: |
Myalgie, artralgie |
|
Časté: | |
|
Celkové poruchy a reakce v místě aplikace | |
|
Velmi časté: |
Únava, zimnice, pyrexie, reakce související s infuzíb |
|
Vyšetření | |
|
Velmi časté |
Snížení tělesné hmotnosti |
a' Preferované termíny, které byly hlášeny v třídě orgánových systémů Infekce a infestace, zahrnují sepsi/septický šok, infekci horních cest dýchacích, herpes zoster a pneumonii. b' Preferované termíny spojené s IRR byly bolest hlavy, vyrážka, bolest zad, zvracení, zimnice, nauzea, dyspnoe, pruritus a kašel.
Neutropenie vedla k odložení dávky u 14 % a 22 % pacientů v populaci u studií fáze 2 a fáze 3, v tomto pořadí.
Při této léčbě může dojít k výskytu závažné a protrahované (>1 týden) neutropenie, což může u pacientů zvýšit riziko rozvinutí závažné infekce. Medián doby trvání neutropenie 3. nebo 4. stupně u populace fáze 2 byl omezený (1 týden); neutropenie 4. stupně trvající >7 dnů se vyskytla u 2 % pacientů. Méně než polovina pacientů v populaci z pivotních studií fáze 2 s neutropenií 3. nebo 4. stupně měla časově související infekce a většina časově souvisejících infekcí byla 1. nebo 2. stupně.
V populaci fáze 3 byla hlášena neutropenie 3. stupně u 22 % pacientů v léčebném rameni s brentuximab vedotinem a neutropenie 4. stupně byla hlášena u 7 % pacientů v léčebném rameni
s brentuximab vedotinem. U žádných pacientů nebylo vyžadováno snížení dávky nebo ukončení léčby z důvodu neutropenie.
V populaci fáze 3 byly hlášeny závažné infekce u 9 % pacientů v léčebném rameni s brentuximab vedotinem. V léčebném rameni s brentuximab vedotinem nebyly hlášeny žádné případy bakterémie, sepse nebo septického šoku.
Periferní senzorická neuropatie vedla k odložení dávky u 13 %, respektive 16 % pacientů v populaci studie fáze 2 a fáze 3. Kromě toho vedly v populaci studie fáze 3 periferní motorická neuropatie a infekce horních cest dýchacích k odložení dávky u 6 % pacientů.
Periferní senzorická neuropatie vedla ke snížení dávek u 9 % a 22 % pacientů v populaci studie ve fázi 2 a fázi 3, v tomto pořadí. Navíc ve fázi 3 vedla ke snížení dávek také periferní motorická neuropatie u 6 % pacientů. Devadesát procent (90 %), respektive šedesát osm procent (68 %) pacientů v populaci fáze 2 a fáze 3 zůstalo během léčby na doporučené dávce 1,8 mg/kg.
Mezi pacienty, u kterých se vyskytla periferní neuropatie v populaci fáze 2, byl medián doby sledování od ukončení léčby do posledního hodnocení přibližně 48,9 týdnů. U 83 % z 89 pacientů, u kterých se vyskytla periferní neuropatie, bylo v době posledního hodnocení zaznamenáno vymizení nebo zlepšení jejich symptomů periferní neuropatie. Medián doby od nástupu do odeznění nebo zlepšení pro všechny příhody činil 16 týdnů (rozmezí od 0,3 týdne do 106,6 týdnů).
Mezi pacienty, u kterých se vyskytla periferní neuropatie v populaci fáze 3, byl medián doby sledování od ukončení léčby do posledního hodnocení přibližně 98 týdnů. U 85 % pacientů, u kterých se vyskytla periferní neuropatie v léčebném rameni s brentuximab vedotinem, bylo v době posledního hodnocení zaznamenáno vymizení nebo zlepšení jejich symptomů periferní neuropatie. Celkově činil medián doby od nástupu do odeznění nebo zlepšení pro příhody periferní neuropatie v léčebném rameni s brentuximab vedotinem 23,4 týdnů (rozmezí od 0,1 týdne do 138,3 týdnů).
Reakce související s infúzí byly nahlášeny u 11%, respektive 15 % pacientů v populaci fáze 2 a fáze
3. Nejčastější nežádoucí účinky spojené s reakcemi souvisejícími s infúzí, ať už v populaci fáze 2, nebo v populaci fáze 3, byly mírné až středně těžké (1. nebo 2. stupně) a zahrnovaly bolest hlavy, vyrážku, bolest zad, zvracení, zimnici, nauzeu, dušnost, svědění a kašel.
Byly hlášeny anafylaktické reakce (viz bod 4.4). K příznakům anafylaktické reakce může patřit, mimo jiné, urtikarie, angioedém, hypotenze a bronchospasmus. Byla hlášena febrilní neutropenie (viz bod 4.2). U jednoho pacienta zařazeného do studie fáze 1 s eskalací dávky se po podání jediné dávky 3,6 mg/kg brentuximab vedotinu vyskytla febrilní neutropenie 5. stupně.
Imunogenita
U pacientů s relabujícím nebo refrakterním HL nebo sALCL ve dvou pivotních studiích fáze 2 byla prováděna vyšetření na přítomnost protilátek proti brentuximab vedotinu každé 3 týdny za použití
senzitivní elektrochemiluminiscenční imunoanalýzy. Pacienti s HL při zvýšeném riziku relapsu nebo progrese po ASCT byli také testováni ve studii fáze 3. U přibližně 7 % pacientů v těchto studiích fáze 2 a u 6 % pacientů ve studii fáze 3 v léčebném rameni s brentuximab vedotinem došlo ke tvorbě perzistentních pozitivních protilátek proti léku (ADA). U dvou pacientů ve studii fáze 2 a dvou pacientů ve studii fáze 3 se vyskytly nežádoucí účinky konzistentní s reakcemi souvisejícími s infuzí, které vedly k přerušení léčby.
Přítomnost protilátek na brentuximab vedotin nekorelovala s klinicky významným snížením hladin brentuximab vedotinu v séru a nevedla ke snížení účinnosti brentuximab vedotinu. I když přítomnost protilátek na brentuximab vedotin nemusí nutně předpovídat rozvoj IRR, u pacientů s přetrvávající ADA pozitivitou byl pozorován vyšší výskyt IRR ve srovnání s pacienty s přechodnou ADA pozitivitou a pacienty, kteří na ADA nikdy pozitivní nebyli.
Opakovaná léčba
Opakovaná léčba přípravkem ADCETRIS byla podána 21 pacientům s relabujícím či refrakterním HL a 8 pacientům s relabujícím sALCL. Medián počtu podaných cyklů byl 7 (rozmezí od 2 do 37) (viz bod 5.1). Typy a hodnocení nežádoucích účinků hlášených u pacientů opakovaně léčených přípravkem ADCETRIS se shodovaly s typy a hodnoceními pozorovanými v kombinovaných pivotních studiích fáze II, s výjimkou periferní motorické neuropatie, u které byl výskyt vyšší (28 % vs. 9 % v pivotních studiích fáze II), a dosahovaly zejména stupně 1 nebo 2. U pacientů byl rovněž pozorován vyšší výskyt artralgie, anémie 3. stupně a bolesti zad než u pacientů sledovaných v kombinovaných pivotních studiích fáze II.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Neexistuje žádné známé antidotum pro předávkování brentuximab vedotinem. V případě předávkování by pacient měl být pečlivě sledován ohledně výskytu nežádoucích účinků, zvláště neutropenie, a má být podána podpůrná léčba (viz bod 4.4).
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika; jiná cytostatika; monoklonální protilátky, ATC kód: L01XC12
Mechanismus účinku
Brentuximab vedotin je konjugát protilátka-lék (Antibody-Drug Conjugate, ADC), z nějž se uvolňuje cytostatikum, které vyvolává apoptotickou buněčnou smrt selektivně v nádorových buňkách exprimujících CD30. Neklinické údaje naznačují, že biologická aktivita brentuximab vedotinu je dána vícekrokovým procesem. Vazba ADC na CD30 na povrchu buňky spouští internalizaci komplexu ADC-CD30, který je poté transportován do lysozomálního kompartmentu. Uvnitř buňky se proteolytickým štěpením uvolňuje jediná definovaná aktivní látka, MMAE. Navázání MMAE na tubulin narušuje síť mikrotubulů v buňce, indukuje zástavu buněčného cyklu a vede k apoptotické smrti nádorové buňky exprimující CD30.
Klasické HL a sALCL exprimují CD30 coby antigen na povrchu maligních buněk. Tato exprese je nezávislá na stádiu choroby, linii terapie či transplantačním stavu. Tyto vlastnosti činí CD30 cílem terapeutické intervence. Vzhledem k mechanismu účinku cílenému na CD30 je brentuximab vedotin schopen překonat chemorezistenci, neboť CD30 je konzistentně exprimován u pacientů, kteří jsou rezistentní vůči kombinované chemoterapii bez ohledu na předchozí transplantační stav. Tento mechanismus účinku brentuximab vedotinu cílený na CD30, konzistentní exprese CD30 u klasických HL a sALCL a terapeutická spektra a klinické důkazy u dvou CD30-pozitivních malignit po vícečetných liniích léčby poskytují biologické zdůvodnění pro jeho použití u pacientů s relabujícím a refraktorním klasickým HL a sALCL po prodělané ASCT nebo bez prodělané ASCT.
Podíl jiných funkcí souvisejících s protilátkou na mechanismu účinku není vyloučen.
Farmakodynamické účinky
Srdeční elektrofyziologie
Z 52 pacientů, kterým bylo podáváno 1,8 mg/kg brentuximab vedotinu každé 3 týdny v rámci jednoramenné, otevřené, multicentrické studie kardiální bezpečnosti fáze 1, bylo hodnotitelných čtyřicet šest (46) pacientů s hematologickými malignitami exprimujícími CD30. Primárním cílem bylo vyhodnotit účinek brentuximab vedotinu na repolarizaci srdečních komor a předem definovanou primární analýzou byla změna QTc od výchozí hodnoty do různých časových bodů během 1. cyklu.
Horní mez 90% intervalu spolehlivosti (CI) pro průměrný účinek na QTc byla <10 ms v každém časovém bodě po výchozím měření v 1. a 3. cyklu. Tyto údaje ukazují na nepřítomnost klinicky významného prodloužení QT intervalu způsobeného brentuximab vedotinem podávaným v dávce
1,8 mg/kg každé 3 týdny u pacientů s malignitami s expresí CD30.
Klinická účinnost a bezpečnost
Hodgkinův lymfom
Studie SG035-0003
Účinnost a bezpečnost brentuximab vedotinu podávaného v monoterapii byla hodnocena v pivotní otevřené, jednoramenné, multicentrické studii u 102 pacientů s relabujícím nebo refrakterním HL. Souhrn výchozích charakteristik pacientů a onemocnění je uveden v tabulce 4 níže.
progrese do 3 měsíců od ukončení úvodní léčby.
nebo refrakterního HL
|
Charakteristiky pacientů |
N = 102 |
|
Medián věku, roky (rozmezí) |
31 let (15-77) |
|
Pohlaví |
48 M (47 %)/54 Ž (53 %) |
|
Výkonnostní stav ECOG | |
|
0 |
42 (41 %) |
|
1 |
60 (59 %) |
|
Předchozí ASCT |
102 (100 %) |
|
Předchozí režimy chemoterapie |
3,5 (1-13) |
|
Doba od ASCT do prvního potransplantačního relapsu |
6,7 měs. (0-131) |
|
Histologicky potvrzené onemocnění s expresí CD30 |
102 (100 %) |
|
Charakteristiky onemocnění | |
|
Primárně refrakterní k úvodní léčběa |
72 (71 %) |
|
Refrakterní k poslední léčbě |
43 (42 %) |
|
B symptomy na začátku léčby |
35 (33 %) |
|
Stádium III při počáteční diagnóze |
27 (26 %) |
|
Stádium IV při počáteční diagnóze |
20 (20 %) |
a Primárně refrakterní HL je definován jako nedosažení kompletní remise při úvodní léčbě nebo
Osmnáct (18) pacientů (18 %) dostalo 16 cyklů brentuximab vedotinu; medián počtu podaných cyklů byl 9 (rozmezí od 1 do 16).
Odpověď na léčbu brentuximab vedotinem byla hodnocena nezávislou komisí (Independent Review Facility, IRF) za použití revidovaných kritérií léčebné odpovědi u maligních lymfomů (Cheson, 2007). Léčebná odpověď byla hodnocena na základě spirálního CT vyšetření hrudníku, krku, břicha a pánve, vyšetření PET a klinických dat. Hodnocení odpovědi byla prováděna v cyklech 2, 4, 7, 10, 13 a 16, s vyšetřením PET v cyklech 4 a 7.
Poměr objektivní odpovědi (ORR) podle hodnocení IRF činil 75 % (76 ze 102 pacientů v populaci intent-to-treat [ITT]) a zmenšení nádoru bylo dosaženo u 94 % pacientů. Kompletní remise (CR) dosáhlo 33 % (34 ze 102 pacientů v ITT populaci). Medián celkového přežití (OS) je 40,5 měsíce (medián doby sledování (doba do úmrtí nebo posledního kontaktu) od první dávky byl 35,1 měsíce (rozmezí 1,8 až 72,9+ měsíce). Odhadovaná celková míra přežití po 5 letech činila 41 % (95 % CI [31 %, 51 %]). Hodnocení prováděná zkoušejícími se většinou shodovala s nezávislým hodnocením snímků. Z léčených pacientů pak 8 pacientů s léčebnou odpovědí podstoupilo alogenní transplantaci kmenových buněk. Další výsledky účinnosti viz tabulka 5.
od první dávky u pacientů, kteří dosáhli objektivní odpovědi (OR) podle IRF, byl 9,0 měsíce. b' Nelze určit.
léčených brentuximab vedotinem v dávce 1,8 mg/kg podávaným každé 3 týdny
|
Nejlepší klinická odpověď (N = 102) |
IRF N (%) |
95% CI |
|
Poměr objektivní odpovědi (CR + PR) |
76 (75) |
64,9; 82,6 |
|
Kompletní remise (CR) |
34 (33) |
24,3; 43,4 |
|
Parciální remise (PR) |
42 (41) |
není k dispozici |
|
Poměr kontroly onemocnění (CR + |
98 (96) |
90,3; 98,9 |
|
PR + SD) | ||
|
Doba trvání odpovědi |
Medián podle IRF |
95% CI |
|
Poměr objektivní odpovědi (CR + PR) a |
6,7 měsíce |
3,6; 14,8 |
|
Kompletní remise (CR) |
27,9 měsíce |
10,8; NEb |
|
Celkové přežití |
95% CI | |
|
Medián |
40,5 měsíce |
28,7; 61,9 |
|
Odhadovaná 5letá míra celk. přežití |
41 % |
31 %, 51 % |
a' Rozmezí doby trvání odpovědi (DOR) bylo 1,2+ měsíce až 43+ měsíce a medián doby sledování
Explorační analýza u jednotlivých pacientů ukázala, že přibližně u 64 % pacientů s HL léčených brentuximab vedotinem v rámci klinické studie SG035-0003 došlo ke zlepšení klinického přínosu na základě delšího přežití bez progrese (PFS) ve srovnání s jejich poslední předchozí linií léčby.
Z 35 pacientů (33 %), kteří měli B symptomy na začátku studie, všechny B symptomy vymizely u 27 pacientů (77 %), a to za medián doby 0,7 měsíce od nasazení brentuximab vedotinu.
Údaje byly získány od pacientů (n = 15) ze studií fáze 1 hodnotících zvyšující se dávky a klinických farmakologických studií a od pacientů (n = 26) z NPP s relabujícím či refraktorním HL, kteří neprodělali ASCT a byli léčeni brentuximab vedotinem v dávce 1,8 mg/kg každé tři týdny.
Počáteční charakteristika pacientů vykazovala selhání několika předchozích chemoterapeutických režimů (medián počtu 3 s rozmezím 1 až 7) před prvním podáním brentuximab vedotinu. Padesát devět procent (59 %) pacientů mělo iniciální diagnózu pokročilého onemocnění (stádium III nebo IV).
Výsledky těchto studií fáze 1 a zkušenosti získané během NPP ukazují, že u pacientů s relabujícím či refraktorním HL, kteří neprodělali ASCT, je možno dosáhnout klinicky významné odpovědi, což prokazuje poměr objektivní odpovědi na léčbu 54 %, hodnoceno zkoušejícím, a poměr kompletní remise 22 % po mediánu 5 cyklů léčby brentuximab vedotinem.
Studie SGN35-005
Účinnost a bezpečnost brentuximab vedotinu byla hodnocena v randomizované, dvojitě zaslepené, placebem kontrolované, dvouramenné multicentrické studii u 329 pacientů s HL při riziku relapsu nebo progrese po ASCT. Ze studie byli vyloučeni pacienti se známým onemocněním mozku / meningeálním onemocněním, včetně PML v anamnéze. Charakteristika pacientů viz tabulka 6.
Z těchto 329 pacientů bylo randomizováno 165 pacientů do léčebného ramene a 164 pacienti byli randomizováni do ramene s placebem. V této studii měli pacienti dostat první dávku po zotavení po ASCT (v rozmezí 30-45 dnů po ASCT). Pacienti byli léčeni přípravkem ADCETRIS v dávce
1,8 mg/kg nebo odpovídajícím placebem intravenózně po dobu 30 minut každé 3 týdny až v 16 cyklech.
Způsobilí pacienti museli mít alespoň jeden z následujících rizikových faktorů:
• HL, který byl refrakterní vůči léčbě první linie,
• Recidivující nebo progresivní HL, který se vyskytl <12 měsíců od konce léčby první linie,
• Extranodální postižení v době recidivy před ASCT včetně extranodálního šíření tkání mízních
uzlin do přilehlých životně důležitých orgánů.
ASCT
|
Charakteristika pacientů |
Brentuximab vedotin N = 165 |
Placebo N = 164 |
|
Medián věku, roky (rozmezí) |
33 let (18-71) |
32 let (18-76) |
|
Pohlaví |
76 M (46 %)/89 Ž (54 |
97 M (59 %)/67 Ž (41 |
|
%) |
%) | |
|
Výkonnostní stav ECOG | ||
|
0 |
87 (53 %) |
97 (59 %) |
|
1 |
77 (47 %) |
67 (41 %) |
|
2 |
1 (1 %) |
0 |
|
Charakteristiky onemocnění | ||
|
Medián počtu předchozích |
2 (2-8) |
2 (2-7) |
|
chemoterapeutických režimů (rozmezí) | ||
|
Medián doby od diagnózy HL po první |
18,7 měs. (6,1-204,0) |
18,8 měs. (7,4-180,8) |
|
dávku (rozmezí) Stadium onemocnění při počáteční diagnóze HL | ||
|
Stadium I |
1 (1 %) |
5 (3 %) |
|
Stadium II |
73 (44 %) |
61 (37 %) |
|
Stadium III |
48 (29 %) |
45 (27 %) |
|
Stadium IV |
43 (26 %) |
51 (31 %) |
|
Není známo |
0 |
2 (1 %) |
|
Stav PET vyšetření před ASCT | ||
|
FDG-AVIDNI |
64 (39 %) |
51 (31 %) |
|
FDG-NEGATIVNÍ |
56 (34 %) |
57 (35 %) |
|
NEPROVEDENO |
45 (27 %) |
56 (34 %) |
|
Extranodální postižení v době recidivy před |
54 (33 %) |
53 (32 %) |
|
ASCT | ||
|
B symptomy3 |
47 (28 %) |
40 (24 %) |
|
Nejlepší odpověď na záchrannou léčbu před ASCTb | ||
|
Kompletní odpověď |
61 (37 %) |
62 (38 %) |
|
Parciální odpověď |
57 (35 %) |
56 (34 %) |
|
Stabilní odpověď |
47 (28 %) |
46 (28 %) |
|
Stav HL po ukončení první linie standardní chemoterapieb | ||
|
Refrakterní |
99 (60 %) |
97 (59 %) |
|
Refrakterní se vyskytl <12 měsíců |
53 (32 %) |
54 (33 %) |
|
Relaps se objevil >=12 měsíců |
13 (8 %) |
13 (8 %) |
a' U refrakterního onemocnění nebo po progresi nebo relapsu po léčbě první linie. b' Stratifikační faktory u randomizace.
Výsledky účinnosti jsou uvedeny v tabulce 7. Primární cílový parametr PFS byl splněn a ukázal rozdíl v mediánu PFS 18,8 měsíců ve prospěch léčebného ramene.
léčených brentuximab vedotinem v dávce 1,8 mg/kg podávaným každé 3 týdny
|
Brentuximab vedotin N = 165 |
Placebo N = 164 |
Poměr stratifikovaných rizik | |
|
Medián podle IRF | |||
|
Přežití bez progrese a |
42,9 měsíců (95% CI [30,4, 42,9]) |
24,1 měsíců (95% CI [11,5, -]) |
0,57 (95% CI [0,40; 0,81]) Stratifikovaný log-rank test P=0,001 |
|
Medián podle zkoušejících | |||
|
Nedosaženo (95% CI [26,4, -]) |
15,8 měsíců (95% CI [8,5, -]) |
0,5 (95% CI [0,36, 0,70])b | |
|
Počet úmrtí (%) | |||
|
Celkové přežití |
28 (17) |
25 (15) |
1,15 (95% CI [0,67, 1,97] |
a' V době primární analýzy byl medián doby sledování pro obě ramena 30 měsíců [rozmezí, 0 až
50].
b' Stratifikovaný log-rank test nebyl proveden pro PFS podle zkoušejících.
Byly provedeny analýzy předem specifikovaných podskupin PFS podle IRF na základě nejlepší odpovědi pacientů na záchrannou terapii před ASCT, stav HL po léčbě první linie, podle věku, pohlaví, výchozí tělesné hmotnosti, výchozím výkonnostním stavu ECOG, počtu léčení před ASCT, zeměpisné oblasti, stavu PET vyšetření před ASCT, stavu B symptomu po selhání léčby první linie a stavu extranodálního onemocnění před ASCT. Analýzy ukázaly konzistentní trend ve prospěch pacientů, kteří dostávali brentuximab vedotin, v porovnání s pacienty, kteří dostávali placebo, s výjimkou pacientů >65 let věku (n=8).
Nebyly pozorovány žádné rozdíly v kvalitě života mezi ramenem s léčebným přípravkem a ramenem s placebem. Analýza využívání zdrojů ve zdravotnictví (Medical resource utilization, MRU) ukázala, že hospitalizace a ambulantní návštěvy, stejně jako pracovní dny / jiné aktivity, které pacienti i pečovatelé zameškali, byly nižší u brentuximab vedotinu v porovnání s placebem u pacientů s HL při zvýšeném riziku relapsu.
Aktualizovaná analýza provedená 3 roky po následné léčbě ukázala neustálé zlepšování PFS (přežití bez progrese) podle IRF (HR = 0,58 [95% CI (0,41; 0,81)]).
Analýzy _post-hoc rizikových _ faktorů
Post-hoc analýzy se prováděly, aby bylo možné vyhodnotit dopad zvýšeného rizika (počet rizikových faktorů) na klinický přínos (tabulka 8). Reprezentativní rizikové faktory pro tyto analýzy byly:
• HL, který se objevil po <12 měsících, nebo HL, který byl refrakterní vůči léčbě první linie,
• Nejlepší odpověď PR nebo SD na poslední záchrannou terapii dle vyšetření CT a/nebo PET,
• Extranodální onemocnění u relapsu před ASCT,
• B symptomy u relapsu před ASCT,
• Dvě nebo více předchozích záchranných terapií.
Výsledky těchto post-hoc analýz naznačují zvýšený klinický přínos pro pacienty se dvěma nebo více rizikovými faktory, ale nenaznačují žádný rozdíl na základě jednotlivých rizikových faktorů.
U pacientů s jedním rizikovým faktorem pro relaps nebo progresi nebyl pozorován žádný přínos ohledem na PFS nebo OS.
Tabulka 8: Shrnutí PFS podle IRF a OS podle počtu rizikových faktorů ve studii fáze 3 u HL po
ASCT
|
Přežití bez progrese podle IRF | ||||||
|
Počet rizikových faktorů |
Počet rizikových faktorů |
Počet rizikových | ||||
|
= 1 |
>2 |
faktorů > 3 | ||||
|
Brentuximab |
Placebo |
Brentuximab |
Placebo |
Brentuximab |
Placebo | |
|
vedotin |
Z II 00 |
vedotin |
N = 136 |
vedotin |
Z II 00 | |
|
N = 21 |
N = 144 |
N = 82 | ||||
|
Počet pacientů s progresí |
9 (43) |
7 (25) |
51 (35) |
68 (50) |
32 (39) |
49 (58) |
|
onemocnění | ||||||
|
nebo úmrtím3 (%) | ||||||
|
Poměr stratifikovan |
1,65 |
0,49 |
0,43 | |||
|
ého rizika |
(95% CI [0.60, 4.55])b |
(95% CI [0.34, 0.71]) |
(95% CI [0.27, 0.68]) | |||
|
Celkové přežití | ||||||
|
Počet rizikových faktorů |
Počet rizikových faktorů |
Počet rizikových | ||||
|
= 1 |
>2 |
faktorů > 3 | ||||
|
Brentuximab |
Placebo |
Brentuximab |
Placebo |
Brentuximab |
Placebo | |
|
vedotin |
Z II 00 |
vedotin |
N = 136 |
vedotin |
Z II 00 | |
|
N = 21 |
N = 144 |
N = 82 | ||||
|
Počet úmrtíc (%) |
5 (24) |
1 (4) |
23 (16) |
24 (18) |
15 (18) |
16 (19) |
|
Poměr stratifikovan |
7,94 |
0,94 |
0,92 | |||
|
ého rizika |
(95% CI [0,93, 68,06])b |
(95% CI [0,53, 1,67]) |
(95% CI [0,45, 1,88]) | |||
a' Úmrtí bez buď předchozí progrese, nebo vice než jedné zmeškané hodnotící návštěvy. b' Naznačuje výsledky z nestratifikované analýzy. c' Případy jsou úmrtí z jakékoliv příčiny.
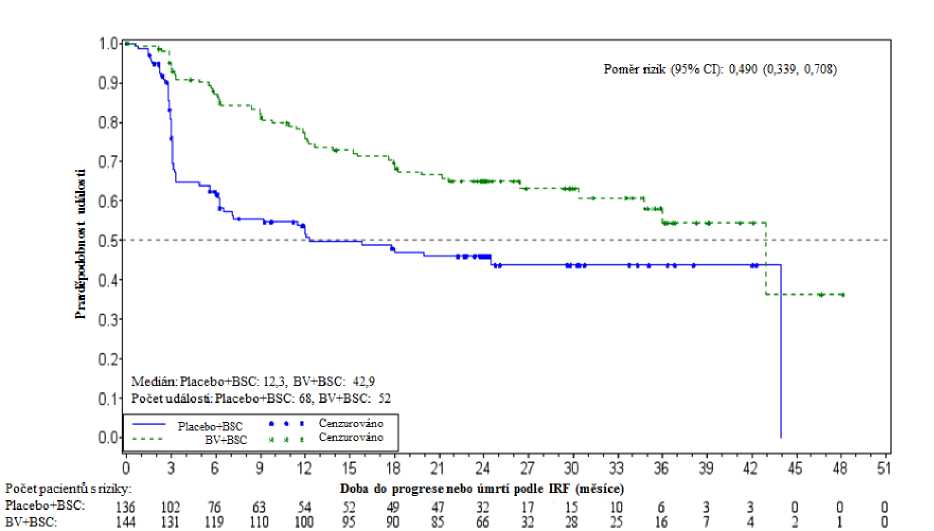
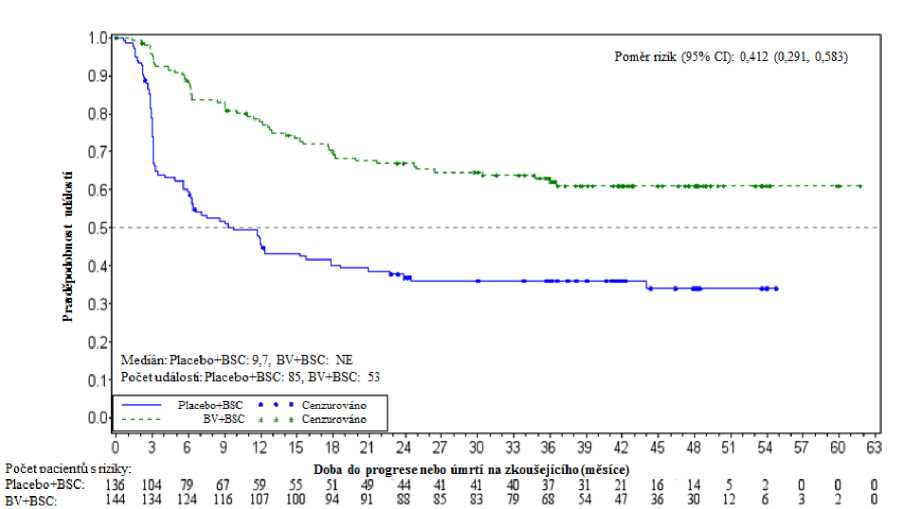
V době aktualizované analýzy (3 roky následného sledování) u pacientů se 2 nebo více rizikovými faktory byl poměr rizik pro PSF podle IRF 0,49 (95% CI [0,34, 0,71]) a poměr rizik pro PFS podle zkoušejících byl 0,41 (95% CI [0,29, 0,58]) (viz obrázky 1 a 2).
Obrázek 1: Kaplan-Meierúv graf PFS podle IRF u pacientů s > 2 rizikovými faktory
Obrázek 2: Kaplan-Meierúv graf PFS na zkoušejícího u pacientů s > 2 rizikovými faktory
Studie SGN35-006 (studie opakované léčby)
Účinnost opakované léčby u pacientů, kteří v minulosti reagovali (CR nebo PR) na léčbu brentuximab vedotinem, byla hodnocena v otevřené multicentrické studii fáze II. Dvaceti pacientům s relabujícím nebo refrakterním HL byla podána počáteční dávka 1,8 mg/kg a jednomu pacientovi počáteční dávka
1,2 mg/kg přípravku ADCETRIS intravenózní infuzí trvající 30 minut každé 3 týdny. Medián počtu podaných cyklů byl 7 (rozmezí od 2 do 37). Z 20 hodnotitelných pacientů s HL opakovaně léčených brentuximab vedotinem dosáhlo 6 pacientů (30 %) kompletní remise a 6 pacientů (30 %) parciální remise, na ORR 60 %. Medián doby trvání odpovědi byl 9,2 a 9,4 měsíce u pacientů, kteří dosáhli OR (CR+PR) a CR, v tomto pořadí.
Systémový anaplastický velkobuněčný lymfom
Studie SG035-0004
Účinnost a bezpečnost brentuximab vedotinu jako jediné účinné látky byla hodnocena v otevřené, jednoramenné, multicentrické studii u 58 pacientů s relabujícím nebo refrakterním sALCL. Shrnutí výchozích charakteristik pacientů a onemocnění viz Tabulka 9 níže.
Tabulka9: Shrnutí výchozích charakteristik pacientů a onemocnění ve studii fáze 2 relabujícího nebo
refrakterního sALCL
|
Charakteristika pacientů |
Z II OO |
|
Medián věku, roky (rozmezí) |
52 let (14-76) |
|
Pohlaví |
33M (57 %)/25Ž (43 %) |
|
Výkonnostní stav ECOGa | |
|
0 |
19 (33 %) |
|
1 |
38 (66 %) |
|
Před ASCT |
15 (26 %) |
|
Předchozí režimy chemoterapie (rozmezí) |
2 (1-6) |
|
Histologicky potvrzené onemocnění s expresí CD30 |
57 (98 %) |
|
Anaplastická lymfomová kináze (ALK)-negativní |
42 (72 %) |
|
onemocnění | |
|
Charakteristika onemocnění | |
|
Primárně refrakterní na léčbu první linieb |
36 (62 %) |
|
Refrakterní na poslední léčbu |
29 (50 %) |
|
Relabující vůči poslední léčbě |
29 (50 %) |
|
Výchozí B symptomy |
17 (29 %) |
|
Stádium III při počáteční diagnóze |
8 (14 %) |
|
Stádium IV při počáteční diagnóze |
21 (36 %) |
a. Jeden pacient měl výchozí stav ECOG 2, což bylo protokolem zakázáno, a je to zachyceno jako Nesplňuje kritéria pro zařazení.
b. Primární refrakterní sALCL je definován jako neschopný dosáhnout kompletní remise vůči nebo s progresí do 3 měsíců po ukončení léčby první linie.
Medián doby od počáteční diagnózy sALCL po první dávku brentuximab vedotinu byl 16,8 měsíců.
Deset (10) pacientů (17 %) dostalo 16 cyklů brentuximab vedotinu; medián počtu podaných cyklů byl 7 (rozmezí 1 až 16).
Odpověď na léčbu brentuximab vedotinem byla hodnocena nezávislou komisí (Independent Review Facility, IRF) za použití revidovaných kritérií léčebné odpovědi u maligních lymfomů (Cheson, 2007). Léčebná odpověď byla hodnocena na základě spirálního CT vyšetření hrudníku, krku, břicha a pánve, vyšetření PET a klinických dat. Hodnocení odpovědi byla prováděna v cyklech 2, 4, 7, 10, 13 a 16, s vyšetřením PET v cyklech 4 a 7.
Poměr objektivní odpovědi (ORR) podle hodnocení IRF činil 86% (50z58 pacientů v populaci intent-to-treat [ITT]) a zmenšení nádoru bylo dosaženo u 97 % pacientů. Odhadovaná 36měsíční doba celkového přežití byla 63 % (medián doby sledování (doba do úmrtí nebo posledního kontaktu) od první dávky byl 33,4 měsíce. Hodnocení prováděná zkoušejícími se většinou shodovala s nezávislým hodnocením snímků. Z léčených pacientů pak 9 pacientů s léčebnou odpovědí podstoupilo alogenní transplantaci kmenových buněk (SCT) a dalších 7 pacientů s léčebnou odpovědí podstoupilo autologní SCT. Další výsledky účinnosti viz tabulka 10.
podáváno 1,8 mg/kg brentuximab vedotinu každé 3týdny
|
Nejlepší klinická odpověď (N = 58) |
IRF N (%) |
95% CI |
|
Poměr objektivní (CR + PR) |
50 (86) |
74,6, 93,9 |
|
Kompletní remise (CR) |
34 (59) |
44,9, 71,4 |
|
Parciální remise (PR) |
16 (28) |
NA |
|
Poměr kontroly onemocnění (CR + PR + SD) |
52 (90) |
78,8, 96,1 |
|
Trvání odpovědi |
Medián podle IRF |
95% CI |
|
Objektivní odpověď (CR + PR)a |
13,2 |
5,7, NEb |
|
Kompletní remise (CR) |
Nedosaženo |
13.0, NE |
|
Celkové přežití |
Medián |
95% CI |
|
Medián |
Nedosaženoc |
21,3, NE |
a' Rozmezí doby trvání odpovědi (DOR) bylo 0,1+ měsíce až 21,7+ měsíce a medián doby sledování
od první dávky u pacientů, kteří dosáhli objektivní odpovědi (OR) podle IRF, byl 11,8 měsíců. b' Nelze určit.
c. Odhadované 36měsíční celkové přežití činilo 63 % (medián doby pozorování (doba po úmrtí nebo poslední kontakt) od první dávky byla 33,4 měsíců).
Explorační analýza u jednotlivých pacientů ukázala, že přibližně u 69 % pacientů s aALCL léčených brentuximab vedotinem v rámci klinické studie SG035-0004 došlo ke zlepšení klinického přínosu na základě delšího přežití bez progrese (PFS) ve srovnání s jejich poslední předchozí linií léčby.
Ze 17 pacientů (29 %), kteří měli B symptomy na začátku studie, u 14 pacientů (82 %) všechny B symptomy vymizely za medián doby 0,7 měsíce od nasazení brentuximab vedotinu.
Studie SGN35-006 (studie opakované léčby)
Účinnost opakované léčby u pacientů, kteří v minulosti reagovali (CR nebo PR) na léčbu brentuximab vedotinem, byla hodnocena v otevřené multicentrické studii fáze 2. Sedmi pacientům s relabujícím sALCL byla podána počáteční dávka 1,8 mg/kg a jednomu pacientovi počáteční dávka 1,2 mg/kg přípravku ADCETRIS intravenózní infuzí trvající 30 minut každé 3 týdny. Medián počtu podaných cyklů byl 8,5 (rozmezí od 2 do 30). Z 8 pacientů s sALCL byli 3 pacienti opakovaně léčeni dvakrát a dostali celkem 11 opakovaných léčeb. Výsledkem opakované léčby brentuximab vedotinem bylo 6 pacientů CR (55 %) a 4 pacienti PR (36 %), na ORR 91 %. Medián doby trvání odpovědi byl 8,8 a 12,3 měsíce u pacientů, kteří dosáhli OR (CR+PR) a CR, v tomto pořadí.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Adcetris u jedné nebo více podskupin pediatrické populace v léčbě Hodgkinova lymfomu a v léčbě anaplastického velkobuněčného lymfomu (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 Farmakokinetické vlastnosti
Farmakokinetika brentuximab vedotinu byla hodnocena ve studiích fáze 1 a v populační farmakokinetické analýze údajů od 314 pacientů. Ve všech klinických hodnoceních byl brentuximab vedotin podáván jako intravenózní infuze.
Maximální koncentrace konjugátu protilátka-lék (ADC) brentuximab vedotinu byly obvykle pozorovány na konci infuze nebo v časovém bodě odběru vzorku nejblíže ke konci infuze. Byl pozorován multiexponenciální pokles sérových koncentrací ADC s terminálním poločasem přibližně
4 až 6 dnů. Hodnoty expozice byly přibližně úměrné dávce. Při více dávkách podávaných každé 3 týdny byla pozorována minimální nebo nebyla pozorována žádná akumulace ADC, což je v souladu s odhadem terminálního poločasu. Typická hodnota Cmax a AUC pro ADC po jednorázové dávce
1.8 mg/kg ve studii fáze 1 činila přibližně 31,98 ^g/ml a 79,41 ^g/ml x den, v tomto pořadí.
Hlavním metabolitem brentuximab vedotinu je MMAE. Medián Cmax, AUC a Tmax pro MMAE po jednorázové dávce 1,8 mg/kg ADC ve studii fáze 1 činil přibližně 4,97 ng/ml, 37,03 ng/ml x den a
2.09 dne, v tomto pořadí. Po více dávkách brentuximab vedotinu se hodnoty expozice MMAE snižovaly; při dalších dávkách byly zaznamenány hodnoty ve výši přibližně 50 % až 80 % hodnoty expozice při první dávce. MMAE je dále metabolizován především na stejně silný metabolit, avšak jeho expozice je o řád nižší, než je expozice MMAE. Proto je nepravděpodobné, že by měl nějaký podstatný přínos k systémovým účinkům MMAE.
V prvním cyklu byla vyšší expozice MMAE spojena se snížením absolutního počtu neutrofilů. Distribuce
Podíl MMAE navázaného na plazmatické proteiny z lidského séra in vitro se pohyboval mezi 68 - 82 %. Není pravděpodobné, že by MMAE vytěsňoval léky vysoce vázané na proteiny nebo byl jimi vytěsňován. MMAE je substrátem P-gp in vitro a při klinických koncentracích není inhibitorem P-gp.
Průměrný distribuční objem v rovnovážném stavu u člověka pro ADC činil přibližně 6 - 10 l. Na základě odhadu populační farmakokinetiky byl typický zdánlivý distribuční objem (VM a VMP) MMAE 7,37 l a 36,4 l, v tomto pořadí.
Biotransformace
Předpokládá se, že ADC je katabolizován jako protein a jeho jednotlivé aminokyseliny jsou recyklovány nebo eliminovány.
Údaje in vivo u zvířat a člověka naznačují, že je metabolizována pouze malá frakce MMAE uvolněného z brentuximab vedotinu. Hladiny metabolitů MMAE v lidské plazmě nebyly měřeny. Bylo prokázáno, že alespoň jeden metabolit MMAE je aktivní in vitro.
MMAE je substrátem CYP3A4 a možná CYP2D6. Údaje in vitro ukazují na to, že metabolismus MMAE, ke kterému dochází, probíhá především prostřednictvím oxidace pomocí CYP3A4/5. Studie in vitro používající lidské jaterní mikrozomy ukazují na to, že MMAE inhibuje pouze CYP3A4/5, a to při mnohem vyšších koncentracích, než byly koncentrace dosažené v rámci klinické aplikace. MMAE neinhibuje jiné izoformy.
MMAE neindukoval žádné hlavní enzymy CYP450 v primárních kulturách lidských hepatocytů. Eliminace
ADC je eliminován katabolismem, s typickými odhadnutými hodnotami CL a poločasu 1,457 l/den a 4-6 dnů, v tomto pořadí.
Eliminace MMAE byla omezena rychlostí jeho uvolňování z ADC, přičemž hodnoty typické zdánlivé CL a poločasu MMAE byly 19,99 l/den a 3-4 dny, v tomto pořadí.
Byla provedena studie exkrece u pacientů, kteří dostali dávku 1,8 mg/kg brentuximab vedotinu. Přibližně 24 % celkového MMAE podaného jako složka ADC během infuze brentuximab vedotinu bylo vyloučeno jak do moči, tak do stolice, v průběhu 1 týdne. Přibližně 72 % vyloučeného MMAE bylo nalezeno ve stolici. Menší množství MMAE (28 %) bylo vyloučeno močí.
Farmakokinetika u zvláštních populací
Populační farmakokinetická analýza prokázala, že výchozí sérová koncentrace albuminu byla významnou kovariancí clearance MMAE. Analýza ukázala, že clearance MMAE byla 2x nižší u pacientů s nízkými koncentracemi albuminu v séru <3,0 g/dl ve srovnání s pacienty s koncentracemi albuminu v séru v normálním rozmezí.
Porucha funkce jater
Studie hodnotila farmakokinetiku brentuximab vedotinu a MMAE po podání 1,2 mg/kg přípravku ADCETRIS pacientům s mírnou (Child-Pugh A; n=1), středně těžkou (Child-Pugh B; n=5) a těžkou (Child-Pugh C; n=1) poruchou funkce jater. Ve srovnání s pacienty s normální funkcí jater se expozice účinkům MMAE u pacientů s poruchou funkce jater zvýšila přibližně 2,3krát (90% CI 1,27-4,12x).
Porucha funkce ledvin
Studie hodnotila farmakokinetiku brentuximab vedotinu a MMAE po podání 1,2 mg/kg přípravku ADCETRIS pacientům s mírnou (n=4), středně těžkou (n=3) a těžkou (n=3) poruchou funkce ledvin. Ve srovnání s pacienty s normální funkcí ledvin se expozice účinkům MMAE u pacientů s těžkou poruchou funkce ledvin (clearance kreatininu < 30 ml/min) zvýšila přibližně 1,9krát (90 % CI 0,85-4,21x). U pacientů s mírnou nebo středně těžkou poruchou funkce ledvin nebyly pozorovány žádné účinky.
Starší pacienti
Klinické studie s brentuximab vedotinem nezahrnovaly dostatečný počet pacientů ve věku 65 let a starších k tomu, aby bylo možné zjistit, zda se jejich odpověď na lék liší od odpovědi u mladších pacientů.
Pediatrická populace
Klinické studie s brentuximab vedotinem nezahrnovaly dostatečný počet pacientů mladších 18 let věku k tomu, aby bylo možné zjistit, zda se jejich farmakokinetický profil liší od farmakokinetického profilu u dospělých pacientů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Mikronukleární studie in vivo s kostní dření potkanů prokázala, že MMAE má aneugenní vlastnosti. Tyto výsledky jsou v souladu s farmakologickým účinkem MMAE na mitotický aparát (narušení mikrotubulové sítě) v buňkách.
Účinky brentuximab vedotinu na mužskou a ženskou fertilitu nebyly studovány. Nicméně výsledky studií toxicity po opakovaném podávání u potkanů naznačují, že brentuximab vedotin má potenciál narušit samčí reprodukční funkci a fertilitu. Testikulární atrofie a degenerace byly částečně reverzibilní po 16týdenním období bez léčby.
Brentuximab vedotin vedl k embryonální/fetální letalitě u březích samic potkanů.
V neklinických studiích byla pozorována lymfoidní deplece a snížená hmotnost thymu, což je konzistentní s farmakologickým narušením mikrotubulů způsobeným MMAE uvolněným z brentuximab vedotinu.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Monohydrát kyseliny citrónové Dihydrát natrium-citrátu Dihydrát trehalosy Polysorbát 80
6.2 Inkompatibility
Studie kompatibility nejsou k dispozici, a proto nesmí být tento léčivý přípravek mísen s jinými léčivými přípravky kromě přípravků uvedených v bodě 6.6.
6.3 Doba použitelnosti
4 roky.
Po rekonstituci/naředění má být přípravek z mikrobiologického hlediska použit okamžitě. Nicméně chemická a fyzikální stabilita po otevření před použitím byla prokázána po dobu 24 hodin při teplotě 2 °C - 8 °C.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2 °C-8 °C).
Chraňte před mrazem.
Uchovávejte injekční lahvičku v původním balení, aby byl přípravek chráněn před světlem.
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci a naředění jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
Injekční lahvička ze skla třídy I s pryžovou butylovou zátkou a hliníkovým/plastovým odtrhovacím uzávěrem, obsahující 50 mg prášku.
Balení s 1 injekční lahvičkou.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Obecná opatření
Je třeba zvážit postupy pro správné zacházení s protinádorovými léky a jejich likvidaci.
Po celou dobu zacházení s tímto léčivým přípravkem je třeba dodržovat správnou aseptickou techniku. Návod na rekonstituci
Obsah jedné injekční lahvičky k jednorázovému použití musí být rekonstituován s 10,5 ml vody na injekci na výslednou koncentraci 5 mg/ml. Jedna injekční lahvička obsahuje 10% navýšení objemu, což poskytuje 55 mg přípravku ADCETRIS v jedné injekční lahvičce a celkový rekonstituovaný objem 11 ml.
1. Nasměrujte proud vody na stěnu injekční lahvičky, nikoli přímo na koláč nebo na prášek.
2. Jemně s injekční lahvičkou zatočte, aby se napomohlo rozpuštění. NEPROTREPÁVEJTE.
3. Rekonstituovaný roztok v lahvičce je čirý až lehce opalescentní, bezbarvý roztok s výsledným pH 6,6.
4. Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není zabarven. V případě přítomnosti zabarvení nebo cizích částic musí být rekonstituovaný roztok zlikvidován.
Příprava infuzního roztoku
Odpovídající množství rekonstituovaného přípravku ADCETRIS je třeba odebrat z injekční lahvičky (lahviček) a přidat do infuzního vaku obsahujícího injekční roztok chloridu sodného 9 mg/ml (0,9%) tak, aby bylo dosaženo výsledné koncentrace 0,4 - 1,2 mg/ml přípravku ADCETRIS. Doporučený objem ředícího roztoku je 150 ml. Již rekonstituovaný ADCETRIS lze též naředit do 5% injekčního roztoku glukózy nebo laktátového Ringerova injekčního roztoku.
Vak jemně obraťte, aby se roztok obsahující ADCETRIS promíchal. NEPROTŘEPÁVEJTE.
Jakékoli množství, které zbyde v lahvičce po natažení objemu na ředění, musí být zlikvidováno v souladu s místními požadavky.
Do připraveného infuzního roztoku přípravku ADCETRIS nebo intravenózního infuzního setu nepřidávejte jiné léčivé přípravky. Po podání by infuzní linka měla být propláchnuta injekčním roztokem chloridu sodného 9 mg/ml (0,9%), 5% injekčním roztokem glukózy nebo laktátovým Ringerovým injekčním roztokem.
Po naředění roztok přípravku ADCETRIS ihned aplikujte infuzí za použití doporučené rychlosti infuze.
Celková doba uchovávání roztoku od rekonstituce do infuze by neměla přesáhnout 24 hodin. Stanovení velikosti dávky:
Výpočet pro stanovení celkové dávky přípravku ADCETRIS (ml) k dalšímu naředění (viz bod 4.2):
Dávka ADCETRISu (mg/kg) x tělesná hmotnost pacienta (kg) Koncentrace rekonstituovaného roztoku v lahvičce (5 mg/ml)
Celková dávka ADCETRISu (ml) k dalšímu naředění
Poznámka: U pacientů s tělesnou hmotností vyšší než 100 kg se má pro výpočet dávky použít 100 kg. Maximální doporučená dávka je 180 mg.
Výpočet pro stanovení celkového potřebného počtu injekčních lahviček přípravku ADCETRIS:
Potřebný počet lahviček ADCETRISu
Celková podávaná dávka ADCETRISu (ml) Celkový objem v jedné lahvičce (10 ml/lahvička)
Tabulka 11: Příklady výpočtů pro pacienty s tělesnou hmotností v rozmezí od 60 kg do 120 kg, kterým
|
je podávána doporučená d |
ávka 1,8 mg/kg přípravku ADCETRIS | ||
|
Hmotnost pacienta (kg) |
Celková dávka = hmotnost pacienta vynásobená doporučenou dávkou f1,8 mg/kgal |
Celkový objem k naředěníb = celková dávka vydělená koncentrací rekonstituovaného roztoku [5 mg/ml] |
Potřebný počet lahviček = celkový objem k naředění vydělený celkovým objemem v jedné lahvičce [10 ml/lahvička] |
|
60 kg |
108 mg |
21,6 ml |
2,16 lahviček |
|
80 kg |
144 mg |
28,8 ml |
2,88 lahviček |
|
100 kg |
180 mg |
36 ml |
3,6 lahviček |
|
120 kgc |
180 mgd |
36 ml |
3,6 lahviček |
a. Pro sníženou dávku pro výpočet použijte 1,2 mg/kg.
b. Ředí se ve 150 ml ředidla a podává se intravenózní infuzí trvající 30 minut každé 3 týdny.
c. U pacientů s tělesnou hmotností vyšší než 100 kg se má pro výpočet dávky použít 100 kg.
d. Maximální doporučená dávka je 180 mg.
Likvidace
ADCETRIS je určen pouze k jednorázovému použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Takeda Pharma A/S Dybendal Alle 10 2630 Taastrup Dánsko
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/12/794/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 25. října 2012
Datum posledního prodloužení registrace: 10. září 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
A. VÝROBCI BIOLOGICKÉ LÉČIVÉ LÁTKY A VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobců biologické léčivé látky
Piramal Healthcare UK Ltd.
Earls Road, Grangemouth Stirlingshire, Scotland FK3 8XG Velká Británie
Lonza AG Lonzastrasse 3930 Visp Švýcarsko
Název a adresa výrobců odpovědných za propouštění šarží
Takeda Austria GmbH St. Peter-StraPe 25 A-4020 Linz Rakousko
Takeda Italia S.p.A.
Via Crosa, 86 28065 Cerano (NO)
Itálie
V příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce odpovědného za propouštění dané šarže.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Je třeba poskytnout další souhrnné údaje o přežití pacientů zařazených do studie SG035-004 včetně subanalýzy pacientů > 100 kg. Data je třeba prezentovat s ohledem na historii provedených kontrol. |
Roční zprávy SG035-004 do roku 2016 nebo jestliže další souhrnné údaje jsou dostatečné (pozorováno nejméně 50 % OS případů) a projeví se dříve. |
|
Je třeba provést neintervenční postmarketingovou studii bezpečnosti (PASS) v obou studovaných populacích pacientů HL a sALCL (n=500) včetně dostatečného počtu pacientů s sALCL (tj. nejméně n=50, studie MA25101). |
Závěrečná zpráva ze studie: 31/12/2018 |
|
Provést jednoramennou studii v obdobné populaci pacientů sALCL se zaměřením na četnost odpovědi, délku trvání odpovědi, četnost (další) ASCT a údaje v subpopulacích (včetně, avšak ne nevyhnutelně omezené na stav ALK a věk) na základě přijatého protokolu CHMP (studie C25006). |
Závěrečná zpráva ze studie: 1. Q. 2021 |
|
Provést jednoramenné sledování r/r HL populace, která nepřichází do úvahy dle ASCT zkoumání četnosti odpovědi, PFS, OS, podíl pacientů -transplantace a bezpečnost (n= cca 60 pacientů) na základě přijatého protokolu CHMP. |
Závěrečná zpráva ze studie: 2. Q. 2017 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU VNĚJŠÍ KRABIČKA
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
ADCETRIS 50 mg prášek pro koncentrát pro infuzní roztok Brentuximabum vedotinum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna injekční lahvička obsahuje brentuximabum vedotinum 50 mg
Po rekonstituci jedna injekční lahvička obsahuje brentuximabum vedotinum 5 mg/ml
3. SEZNAM POMOCNÝCH LÁTEK
Pomocné látky: Monohydrát kyseliny citronové, dihydrát natrium-citrátu, dihydrát trehalosy, polysorbát 80
Dále viz příbalová informace
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro koncentrát pro infuzní roztok. 1 injekční lahvička
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Pro intravenózní podání po rekonstituci a naředění Přečtěte si příbalovou informaci
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v chladničce Chraňte před mrazem
Uchovávejte injekční lahvičku v původním balení, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Pouze pro jednorázové použití
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Takeda Pharma A/S Dybendal Alle 10 2630 Taastrup Dánsko
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/12/794/001
13. ČÍSLO ŠARŽE
Lot
14. KLASIFIKACE PRO VÝDEJ
Výdej léčivého přípravku vázán na lékařský předpis
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Nevyžaduje se - odůvodnění přijato
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OBALU OZNAČENÍ NA INJEKČNÍ LAHVIČCE
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU A CESTA/CESTY PODÁNÍ
ADCETRIS 50 mg prášek pro koncentrát pro infuzní roztok Brentuximabum vedotinum i.v. podání
2. ZPŮSOB PODÁNÍ
Pro intravenózní podání po rekonstituci a naředění
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
50 mg
6. JINÉ
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Adcetris 50 mg prášek pro koncentrát pro infuzní roztok brentuximabum vedotinum
'V Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Adcetris a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete Adcetris používat
3. Jak se Adcetris podává
4. Možné nežádoucí účinky
5. Jak Adcetris uchovávat
6. Obsah balení a další informace
1. Co je Adcetris a k čemu se používá
Adcetris obsahuje léčivou látku brentuximab vedotin, protinádorový lék, který se skládá z monoklonální protilátky navázané na látku určenou k ničení nádorových buněk. Monoklonální protilátka tuto látku dopravuje do nádorových buněk. Monoklonální protilátka je bílkovina, která rozpoznává určité nádorové buňky.
Adcetris se používá k léčbě klasického Hodgkinova lymfomu, který:
- se vrátil po infuzi Vašich vlastních zdravých kmenových buněk do Vašeho těla (autologní transplantace kmenových buněk) nebo na ni nereagoval; nebo
- se vrátil po alespoň dvou předchozích protinádorových terapiích nebo na ně vůbec nereagoval, a další kombinovaná protinádorová terapie již není ve Vašem případě možná nebo není možné, abyste podstoupil(a) autologní transplantaci kmenových buněk.
Klasický Hodgkinův lymfom vykazuje na buněčném povrchu specifické bílkoviny, které se liší od neklasického Hodgkinova lymfomu.
Adcetris se také používá ke snížení pravděpodobnosti návratu klasického Hodgkinova lymfomu po autologní transplantaci kmenových buněk u pacientů s určitými rizikovými faktory.
Adcetris se používá k léčbě systémového anaplastického velkobuněčného lymfomu, který se nachází v lymfatických uzlinách a/nebo jiných částech Vašeho těla, který:
- nereagoval na jiné typy protinádorové léčby nebo
- se vrátil po předchozí protinádorové léčbě.
Jak Hodgkinův lymfom, tak systémový anaplastický velkobuněčný lymfom jsou druhy nádorového onemocnění bílých krvinek.
NEPOUŽÍVEJTE Adcetris, jestliže:
- jste alergický(á) na brentuximab vedotin nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- v současné době používáte bleomycin, protinádorový lék.
Upozornění a opatření
Při prvním podání tohoto přípravku a během průběhu léčby svého lékaře informujte, jestliže:
- se u Vás vyskytne zmatenost, poruchy myšlení, ztráta paměti, rozmazané vidění nebo ztráta zraku, snížená síla, snížená schopnost ovládání nebo čití v jedné horní nebo dolní končetině, změna způsobu chůze nebo ztráta rovnováhy, jelikož to mohou být příznaky závažného a potenciálně smrtelného onemocnění mozku známého jako progresivní multifokální leukoencefalopatie (PML). Pokud tyto příznaky máte před zahájením léčby tímto přípravkem, informujte ihned svého lékaře o jakýchkoli změnách těchto příznaků. Informujte také o Vaší léčbě svého partnera/partnerku nebo ošetřovatele, protože si mohou všimnout příznaků, kterých si nejste vědom(a).
- trpíte silnou a přetrvávající bolestí břicha s pocitem na zvracení a zvracením či bez nich, protože se může jednat o příznaky závažného a potenciálně smrtelného onemocnění známého jako pankreatitida (zánět slinivky břišní).
- trpíte novou nebo zhoršující se dušností nebo kašlem, protože se může jednat o příznaky závažné plicní komplikace (plicní toxicity), která může vést k úmrtí.
- používáte nebo jste v minulosti užíval(a) léky, které mohou působit na imunitní systém, jako např. chemoterapeutika nebo imunosupresiva.
- máte nebo se domníváte, že máte, infekci. Některé infekce mohou být závažné a mohou být způsobeny viry, bakteriemi nebo jinými příčinami, které mohou být život ohrožující.
- se u Vás objeví hvízdavý zvuk při dýchání (sípání) / dýchací potíže, kopřivka, svědění nebo otok (známky reakce na infuzi). Podrobnější informace viz „Reakce na infuzi“ v bodě 4.
- máte jakékoli potíže spojené se změnou citlivosti kůže, zejména na rukou nebo chodidlech, jako např. znecitlivění, mravenčení a pocit pálení, bolest, nepříjemný pocit nebo slabost (neuropatie).
- máte bolesti hlavy, cítíte se unavený(á), máte závratě, bledou pokožku (anémie) nebo neobvyklé krvácení nebo tvorba modřin, krvácení po odběru krve, které trvá déle než obvykle, nebo krvácení z dásní (trombocytopenie).
- se u Vás objeví zimnice nebo třesavka nebo je Vám horko; změřte si teplotu, protože byste mohl(a) mít horečku. Horečka spolu s nízkým počtem bílých krvinek může být známkou závažné infekce.
- se u Vás vyskytnou závratě, snížená tvorba moči, zmatenost, zvracení, pocit na zvracení, otok, dušnost nebo poruchy srdečního rytmu (může se jednat o potenciálně život ohrožující komplikaci známou jako syndrom nádorového rozpadu).
- zaznamenáte příznaky podobné chřipce následované bolestivou červenou nebo nafialovělou vyrážkou, která se šíří a vytváří puchýře, včetně rozsáhlého odlučování kůže, které může být život ohrožující (může se jednat o závažnou kožní reakci známou jako Stevens-Johnsonův syndrom a toxickou epidermální nekrolýzu).
- zaznamenáte novou nebo zhoršující se bolest žaludku, pocit na zvracení, zvracení, zácpu, protože se může jednat o příznaky závažné žaludeční a střevní komplikace (gastrointestinální komplikace), která může vést k úmrtí.
- máte abnormální výsledky jaterních testů, což může souviset se závažným poškozením jater (hepatotoxicitou), které může vést k úmrtí. Onemocnění jater a další zdravotní potíže, které mohly existovat předtím, než jste začal(a) užívat přípravek Adcetris, a některé léky, které užíváte v současné době, mohou zvýšit riziko poškození jater.
- se cítíte unavený(á), často močíte, máte zvýšený pocit žízně, zvýšenou chuť k jídlu s neúmyslnou ztrátou tělesné hmotnosti nebo trpíte podrážděností (hyperglykemie).
- máte potíže s ledvinami nebo játry.
Váš lékař bude provádět pravidelná vyšetření krve za účelem zajistit, že Vám tento přípravek může být bezpečně podáván.
Další léčivé přípravky a Adcetris
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a), nebo pokud začnete užívat nové léky. A to včetně rostlinných léků a jiných léků, které jsou dostupné bez lékařského předpisu.
Těhotenství, kojení a plodnost
Během Vaší léčby tímto přípravkem musíte Vy a Váš partner / Vaše partnerka používat dvě metody účinné antikoncepce. Ženy musí v používání antikoncepce pokračovat po dobu 6 měsíců po poslední dávce přípravku Adcetris.
Pokud jste těhotná, používejte tento přípravek pouze tehdy, pokud jste Vy a Váš lékař dospěli k rozhodnutí, že přínos pro Vás převažuje nad potenciálním rizikem pro nenarozené dítě.
Je důležité, abyste před léčbou a během ní svého lékaře informovala, pokud jste těhotná, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět.
Pokud kojíte, poraďte se se svým lékařem, zda by Vám tento přípravek měl být podáván.
Mužům léčeným tímto lékem se doporučuje nechat si před zahájením léčby zmrazit a uložit vzorky spermatu. Mužům se nedoporučuje počít dítě během léčby tímto přípravkem a po dobu 6 měsíců po poslední dávce přípravku.
Řízení dopravních prostředků a obsluha strojů
Vaše léčba může mít vliv na Vaši schopnost řídit a obsluhovat stroje. Pokud se během léčby necítíte dobře, neřiďte a neobsluhujte stroje.
Adcetris obsahuje sodík
Tento přípravek obsahuje maximálně 2,1 mmol (tj. 47 mg) sodíku v jedné dávce. To je nutné vzít v úvahu u pacientů dodržujících dietu s omezeným příjmem sodíku.
3. Jak se Adcetris podává
Máte-li jakékoli otázky týkající se používání tohoto přípravku, zeptejte se lékaře nebo zdravotní sestry, kteří Vám podávají infuzi.
Dávka a frekvence
Dávka tohoto přípravku závisí na Vaší tělesné hmotnosti. Obvyklá počáteční dávka přípravku Adcetris je 1,8 mg/kg, podávaná jednou za 3 týdny po dobu nepřesahující jeden rok. Pokud trpíte poruchou funkce ledvin nebo jater, Váš lékař může snížit Vaši počáteční dávku na 1,2 mg/kg.
Adcetris se má podávat pouze dospělým. Není určen k použití u dětí.
Jak se Adcetris podává
Tento přípravek Vám bude podáván do žíly (intravenózně) jako infuze. Bude Vám podán Vaším lékařem nebo zdravotní sestrou v průběhu 30 minut. Váš lékař nebo zdravotní sestra na Vás budou dohlížet během a po ukončení infuze.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Reakce na infuzi
Léky tohoto typu (monoklonální protilátky) mohou vyvolat reakce na infuzi, jako jsou např.:
- vyrážka
- dušnost
- dýchací potíže
- kašel
- svírání na hrudi
- horečka
- bolest zad
- zimnice
- bolest hlavy
- nevolnosti (pocit na zvracení) nebo zvracení.
Reakce na infuzi tohoto přípravku postihují více než 1 osobu z 10.
Tyto typy reakcí se většinou objeví do několika minut až několika hodin po ukončení infuze. Mohou se však rozvinout za více než několik hodin po ukončení infuze, i když to je méně časté. Tyto reakce na infuzi mohou být závažné nebo i smrtelné (známé jako anafylaktická reakce). Není známo, jak často jsou reakce na infuzi tohoto léčivého přípravku závažné nebo smrtelné.
Mohou Vám být podány další léky, jako např.
- antihistaminika, kortikosteroidy nebo paracetamol,
ke zmírnění některých z výše uvedených reakcí, pokud se u Vás již takové reakce při podání tohoto typu přípravku vyskytly.
Pokud se domníváte, že se u Vás v minulosti vyskytla podobná reakce, sdělte to svému lékaři PŘEDTÍM, než Vám bude tento přípravek podán.
Pokud se u Vás vyskytnou reakce na infuzi (jak je uvedeno výše), Váš lékař podávání tohoto přípravku zastaví a zahájí podpůrnou léčbu.
Pokud bude Vaše infuze znovu zahájena, Váš lékař může prodloužit dobu, během které je Vám infuze podávána, abyste ji mohl(a) lépe snášet.
Ihned informujte svého lékaře, pokud si všimnete jakýchkoli z následujících příznaků, protože některé z nich mohou být známkami závažného onemocnění, které může vést k úmrtí:
- příznaky progresivní multifokální leukoencefalopatie (PML) jako např. zmatenost, poruchy myšlení, ztráta paměti, rozmazané vidění nebo ztráta zraku, snížená síla, snížená schopnost ovládání nebo čití v jedné horní nebo dolní končetině, změna způsobu chůze nebo ztráta rovnováhy (podrobnější informace viz bod 2). Frekvenci tohoto onemocnění nelze z dostupných údajů určit.
- příznaky zánětu slinivky břišní (pankreatitida), jako je silná a přetrvávající bolest břicha s pocitem na zvracení a zvracením či bez nich (postihuje méně než 1 osobu ze 100).
- dušnost nebo kašel (postihují více než 1 osobu z 10)
- příznaky podobné chřipce následované bolestivou červenou nebo nafialovělou vyrážkou, která se šíří a vytváří puchýře, včetně rozsáhlého odlučování kůže (postihuje méně než 1 osobu
z 1000).
- změna vnímání nebo citlivosti, zejména na kůži, necitlivost, mravenčení, nepříjemný pocit a pocit pálení, slabost nebo bolest v rukou nebo nohou (neuropatie, postihuje více než 1 osobu z 10)
- pocit slabosti (postihuje více než 1 osobu z 10)
- zácpa (postihuje více než 1 osobu z 10)
- průjem, zvracení (postihuje více než 1 osobu z 10)
- zimnice nebo třesavka (postihuje více než 1 osobu z 10)
- pocit únavy, časté močení, zvýšený pocit žízně, zvýšená chuť k jídlu s neúmyslnou ztrátou tělesné hmotnosti a podrážděnost (mohou to být známky hyperglykemie, která postihuje méně než 1 osobu z 10)
- neobvyklé krvácení nebo tvorba modřin, krvácení po odběru krve, které trvá déle než obvykle, nebo krvácení z dásní (mohou to být známky trombocytopenie, která postihuje méně než
1 osobu z 10)
- bolesti hlavy, máte závratě, bledou pokožku (mohou to být známky anémie, která postihuje méně než 1 osobu z 10)
Mohou se u Vás vyskytnout následující nežádoucí účinky:
Velmi časté nežádoucí účinky (postihují více než 1 osobu z 10)
- snížený počet bílých krvinek
- infekce horních cest dýchacích
- snížení tělesné hmotnosti
- infekce
- pocit na zvracení
- bolest břicha
- svědění
- neobvyklé vypadávání nebo řídnutí vlasů
- svalová bolest
- bolest kloubů nebo bolestivé, oteklé klouby
Časté nežádoucí účinky (postihují méně než 1 osobu z 10)
- otrava krve (sepse) a/nebo septický šok (život ohrožující druh sepse); pneumonie
- snížený počet krevních destiček
- závrať
- puchýře, na kterých se mohou vytvářet krusty nebo strupy
- zvýšená hladina cukru v krvi
- zvýšení hodnot jaterních enzymů
Méně časté nežádoucí účinky (postihují méně než 1 osobu ze 100)
- Syndrom nádorového rozpadu - potenciálně život ohrožující stav, při kterém se u Vás mohou vyskytnout závratě, snížená tvorba moči, zmatenost, zvracení, pocit na zvracení, otok, dušnost nebo poruchy srdečního rytmu.
- bolestivé, krémově žluté, vyvýšené skvrny v ústech (moučnivka)
Vzácné nežádoucí účinky (postihují méně než 1 osobu z 1000)
- Stevens-Johnsonův syndrom a toxická epidermální nekrolýza - vzácné, závažné onemocnění, při kterém se u Vás mohou objevit příznaky podobné chřipce následované bolestivou červenou nebo nafialovělou vyrážkou, která se šíří a vytváří puchýře, včetně rozsáhlého odlučování kůže.
Není známo (frekvenci z dostupných údajů nelze určit)
- snížená hladina bílých krvinek s horečkou
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na štítku injekční lahvičky a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Neotevřená injekční lahvička: Uchovávejte v chladničce (2 °C - 8 °C). Chraňte před mrazem. Uchovávejte injekční lahvičku v původním obalu, aby byl přípravek chráněn před světlem.
Rekonstituovaný/ naředěný roztok: Použijte okamžitě nebo uchovávejte v chladničce (2 °C - 8 °C) a použijte do 24 hodin.
Nepoužívejte tento přípravek, pokud si před podáním všimnete jakýchkoli cizích částic nebo nežádoucího zabarvení.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Tento přípravek zlikviduje lékař nebo zdravotní sestra. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co Adcetris obsahuje
- Léčivou látkou je brentuximabum vedotinum. Jedna injekční lahvička obsahuje brentuximabum vedotinum 50 mg. Po rekonstituci jeden ml roztoku obsahuje 5 mg přípravku Adcetris.
- Dalšími složkami jsou monohydrát kyseliny citronové, dihydrát natrium-citrátu, dihydrát trehalosy a polysorbát 80. Další informace týkající se sodíku viz bod 2.
Jak Adcetris vypadá a co obsahuje toto balení
Adcetris je bílý až téměř bílý koláč nebo prášek pro koncentrát pro infuzní roztok dodávaný ve skleněné injekční lahvičce.
Jedno balení přípravku Adcetris obsahuje jednu injekční lahvičku.
Držitel rozhodnutí o registraci
Takeda Pharma A/S Dybendal Alle 10 2630 Taastrup Dánsko
Výrobce
Takeda Austria GmbH St. Peter-StraPe 25 A-4020 Linz Rakousko
Takeda Italia S.p.A.
Via Crosa, 86 Cerano, 28065 Itálie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
|
Belgie/Belgique/Belgien Takeda Belgium Tel/Tél: +32 2 464 06 11 takeda-belgium@takeda.com |
Lietuva Takeda, UAB Tel: +370 521 09 070 |
|
BtnrapHH TaKega Etnrapna Ten.: + 359 2 958 27 36; + 359 2 958 15 29 |
Luxembourg/Luxemburg Takeda Belgium Tel/Tél: +32 2 464 06 11 takeda-belgium@takeda.com |
|
Česká republika Takeda Pharmaceuticals Czech Republic s.r.o. Tel: + 420 234 722 722 |
Magyarország Takeda Pharma Kft. Tel: +361 2707030 |
|
Danmark Takeda Pharma A/S Tlf: +45 46 77 11 11 |
Malta Takeda Italia S.p.A. Tel: +39 06 502601 |
|
Deutschland Takeda GmbH Tel: 0800 825 3325 medinfo@takeda.de |
Nederland Takeda Nederland bv Tel: +31 23 56 68 777 nl.medical.info@takeda.com |
|
Eesti Takeda Pharma AS Tel: +372 6177 669 |
Norge Takeda Nycomed AS Tlf: +47 6676 3030 infonorge@takeda.com |
|
ElláSa TAKEDA EAAAI A.E Tel: +30 210 6729570 gr.info@takeda.com |
Osterreich Takeda Pharma Ges.m.b.H. Tel: +43 (0) 800-20 80 50 |
|
Espaňa Takeda Farmacéutica Espana S.A Tel: +34 917 14 99 00 spain@takeda.com |
Polska Takeda Polska Sp. z o.o tel. + 48 22 608 13 00 |
|
France Takeda France Tel. +33 1 46 25 16 16 |
Portugal Takeda Farmaceuticos Portugal, Lda. Tel: + 351 21 120 1457 |
|
Hrvatska Takeda Pharmaceuticals Croatia d.o.o. Tel: +385 1 377 88 96 |
Románia Takeda Pharmaceuticals SRL Tel: +40 21 335 03 91 |
|
Ireland Takeda Products Ireland Limited Tel: +44 (0)1628 537 900 |
Slovenija Takeda GmbH, Podružnica Slovenija Tel: + 386 (0) 59 082 480 |
|
Ísland Vistor hf. tel: +354 535 7000 vistor@vistor.is |
Slovenská republika Takeda Pharmaceuticals Slovakia s.r.o Tel: +421 (2) 20 602 600 |
Suomi/Finland
Italia
Takeda Italia S.p.A. Tel: +39 06 502601
Takeda Oy
Tel. +358 20 746 5000 infoposti@takeda.com
Kúrcpog
Takeda Pharma A/S T^: +45 46 77 11 11
Sverige
Takeda Pharma AB Tel: +46 8 731 28 00 infosweden@takeda.com
Latvija
Takeda Latvia SIA Tel: +371 67840082
United Kingdom
Takeda UK Ltd
Tel: +44 (0)1628 537 900
Tato příbalová informace byla naposledy revidována
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky: http://www.ema.europa.eu/
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Návod k rekonstituci
Obsah jedné injekční lahvičky k jednorázovému použití musí být rekonstituován s 10,5 ml vody na injekci na výslednou koncentraci 5 mg/ml. Jedna injekční lahvička obsahuje 10% navýšení objemu, což poskytuje 55 mg přípravku Adcetris v jedné injekční lahvičce a celkový rekonstituovaný objem 11 ml.
1. Nasměrujte proud vody na stěnu injekční lahvičky, nikoli přímo na koláč nebo prášek.
2. Jemně s injekční lahvičkou zatočte, aby se napomohlo rozpuštění. NEPROTREPÁVEJTE.
3. Rekonstituovaný roztok v lahvičce je čirý až lehce opalescentní, bezbarvý roztok s výsledným pH 6,6.
4. Rekonstituovaný roztok je třeba vizuálně zkontrolovat, zda neobsahuje cizí částice nebo není zabarven. V případě přítomnosti zabarvení nebo cizích částic musí být rekonstituovaný roztok zlikvidován.
Příprava infuzního roztoku
Odpovídající množství rekonstituovaného přípravku Adcetris je třeba odebrat z injekční lahvičky (lahviček) a přidat do infuzního vaku obsahujícího injekční roztok chloridu sodného 9 mg/ml (0,9%) tak, aby bylo dosaženo výsledné koncentrace 0,4 - 1,2 mg/ml přípravku Adcetris.
Doporučený objem ředidla je 150 ml.
Již rekonstituovaný Adcetris lze též naředit do 5% injekčního roztoku glukózy nebo laktátového Ringerova injekčního roztoku.
Vak jemně obraťte, aby se roztok obsahující Adcetris promíchal. NEPROTREPÁVEJTE.
Jakékoli množství, které zbude v lahvičce po natažení objemu na ředění, musí být zlikvidováno v souladu s místními požadavky.
Do připraveného infuzního roztoku přípravku Adcetris nebo intravenózního infuzního setu nepřidávejte jiné léčivé přípravky. Po podání má být infuzní linka propláchnuta injekčním roztokem chloridu sodného 9 mg/ml (0,9%), 5% injekčním roztokem glukózy nebo laktátovým Ringerovým injekčním roztokem.
Po naředění roztok přípravku Adcetris ihned aplikujte infuzí za použití doporučené rychlosti infuze. Celková doba uchovávání roztoku od rekonstituce do infuze nemá přesáhnout 24 hodin.
Likvidace
Adcetris je určen pouze k jednorázovému použití.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
44