Abraxane 5 Mg/Ml
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Abraxane 5 mg/ml, prášek pro přípravu infuzní suspenze.
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje paclitaxelum 100 mg ve formě nanočástic vázaných na albumin. Jedna injekční lahvička obsahuje paclitaxelum 250 mg ve formě nanočástic vázaných na albumin.
Jeden ml suspenze po rekonstituci obsahuje paclitaxelum 5 mg ve formě nanočástic vázaných na albumin.
Pomocné látky se známým účinkem
Jeden ml koncentrátu obsahuje 0,183 mmol sodíku, což je 4,2 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro přípravu infuzní suspenze.
Rekonstituovaná suspenze má pH 6-7,5 a osmolalitu 300-360 mOsm/kg. Prášek je bílý až žlutý.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Abraxane v monoterapii je indikován pro léčbu metastazujícího karcinomu prsu u dospělých pacientů, u kterých selhala první linie léčby metastazujícího onemocnění, a pro pacienty, pro něž není standardní léčba obsahující antracykliny indikována (viz bod 4.4).
Abraxane v kombinaci s gemcitabinem je indikován jako lék první linie pro léčbu dospělých pacientů s metastazujícím karcinomem pankreatu.
Abraxane v kombinaci s karboplatinou je indikován jako lék první linie k léčbě nemalobuněčného karcinomu plic u dospělých pacientů, kteří nejsou kandidáty na potenciálně kurativní chirurgický zákrok a/nebo radiační terapii.
4.2 Dávkování a způsob podání
Abraxane smí být podáván pouze pod dohledem kvalifikovaného onkologa na pracovištích, která se specializují na podávání cytostatických přípravků. Neměl by nahrazovat jiné formy paklitaxelu a ani by jimi neměl být nahrazován.
Dávkování
Karcinom _ prsu
Doporučená dávka přípravku Abraxane je 260 mg/m2 podávaná nitrožilně po dobu 30 minut každé 3 týdny.
Úpravy dávkování během léčby karcinomu prsu
Pacientům, u nichž se rozvine závažná neutropenie (počet neutrofilů <500 buněk/mm3 po dobu týdne nebo déle) nebo závažná senzorická neuropatie během léčby přípravkem Abraxane, by mělo být dávkování v dalších cyklech sníženo na 220 mg/m2. Po opětovném výskytu závažné neutropenie nebo závažné senzorické neuropatie by mělo být provedeno další snížení dávky na 180 mg/m2. Abraxane nesmí být podáván, dokud se počet neutrofilů nezvýší na >1 500 buněk/mm3. U senzorické neuropatie 3. stupně je třeba pozastavit léčbu až do návratu na stupeň 1 nebo 2 a ve všech dalších cyklech pokračovat v podávání snížené dávky.
Karcinom pankreatu
Doporučená dávka přípravku Abraxane v kombinaci s gemcitabinem je 125 mg/m2 podávaná nitrožilně po dobu 30 minut 1., 8. a 15. den každého 28denního cyklu. Doporučená dávka souběžně podávaného gemcitabinu je 1 000 mg/m2 podávaná nitrožilně po dobu 30 minut ihned po ukončení podávání přípravku Abraxane 1., 8. a 15. den každého 28denního cyklu.
Úpravy dávky v průběhu léčby karcinomu pankreatu
Tabulka 1: Úrovně snížení dávky u pacientů s karcinomem pankreatu
|
Hladina dávky |
Dávka přípravku Abraxane (mg/m2) |
Dávka gemcitabinu (mg/m2) |
|
Celá dávka |
125 |
1 000 |
|
1. úroveň snížení dávky |
100 |
800 |
|
2. úroveň snížení dávky |
75 |
600 |
|
Pokud je nutné další snížení dávky |
Ukončete léčbu |
Ukončete léčbu |
Tabulka 2: Úpravy dávkování při neutropenii a/nebo trombocytopenii na začátku cyklu nebo v průběhu cyklu u pacientů s karcinomem pankreatu
|
Den cyklu |
Počet ANC (buňky/mm3) |
Počet trombocytů (buňky/mm3) |
Dávka přípravku Abraxane |
Dávka gemcitabinu | |
|
1. den |
<1 500 |
NEBO |
<100 000 |
Odložte podávání do obnovení počtu buněk | |
|
8. den |
>500, ale <1 000 |
NEBO |
>50 000, ale <75 000 |
Snižte dávky o 1 úroveň | |
|
<500 |
NEBO |
<50 000 |
Nepodávejte dávky | ||
|
15. den: Pokud byly dávky 8. den podány beze změny: | |||||
|
15. den |
>500, ale <1 000 |
NEBO |
>50 000, ale <75 000 |
Podejte dávku stejné úrovně, jako byla podána 8. den, a následně podejte leukocytární růstové faktory NEBO Snižte dávky o 1 úroveň oproti dávkám, které byly podány 8. den | |
|
<500 |
NEBO |
<50 000 |
Nepodávejte dávky | ||
|
15. den: Pokud byly dávky podané 8. den snížené: | |||||
|
15. den |
>1 000 |
A |
>75 000 |
Vraťte se k úrovni dávek z 1. dne a následně podejte leukocytární růstové faktory NEBO | |
|
Podejte stejné dávky jako 8. den | ||||
|
>500, ale <1 000 |
NEBO |
>50 000, ale <75 000 |
Podejte dávku stejné úrovně, jako byla podána 8. den, a následně podejte leukocytární růstové faktory NEBO Snižte dávky o 1 úroveň oproti dávkám, které byly podány 8. den | |
|
<500 |
NEBO |
<50 000 |
Nepodávejte dávky | |
|
15. den: Pokud dávky nebyly 8. den podány: | ||||
|
15. den |
>1 000 |
A |
>75 000 |
Vraťte se k úrovni dávek z 1. dne a následně podejte leukocytární růstové faktory NEBO Snižte dávky o 1 úroveň oproti dávkám, které byly podány 1. den |
|
>500, ale <1 000 |
NEBO |
>50 000, ale <75 000 |
Snižte dávky o 1 úroveň a následně podejte leukocytární růstové faktory NEBO Snižte dávky o 2 úrovně oproti dávkám z 1. dne | |
|
<500 |
NEBO |
<50 000 |
Nepodávejte dávky | |
Zkratky: absolutní počet neutrofilních granulocytů (ANC = Absolute Neutrophil Count); leukocyty (WBC =
white blood cells)
Tabulka 3: Úpravy dávkování z důvodu dalších nežádoucích účinků u pacientů s karcinomem pankreatu___
|
Nežádoucí účinek |
Dávka přípravku Abraxane |
Dávka gemcitabinu |
|
Febrilní neutropenie: Stupeň 3 nebo 4 |
Pozastavte podávání dávek, dokud neustoupí horečka a ANC nebude >1 500; pokračujte na nejbližší nižší úrovni dáveka | |
|
Periferní neuropatie: Stupeň 3 nebo 4 |
Nepodávejte dávku, dokud nedojde ke zlepšení na stupeň <1; pokračujte na nejbližší nižší úrovni dáveka |
Podejte stejnou dávku |
|
Kožní toxicita: Stupeň 2 nebo 3 |
Pokračujte na nejbližší nižší úrovni dáveka; pokud nežádoucí účinek přetrvává, ukončete léčbu | |
|
Gastrointestinální toxicita: Mukositida 3. stupně nebo průjem 3. stupně |
Nepodávejte dávky, dokud nedojde ke zlepšení na stupeň <1; pokračujte na nejbližší nižší úrovni dáveka | |
aÚrovně snížení dávek viz tabulka 1
Nemalobuněčný karcinom _plic
Doporučená dávka přípravku Abraxane je 100 mg/m2 podávaná nitrožilní infuzí po dobu 30 minut 1., 8. a 15. den každého 21denního cyklu. Doporučená dávka karboplatiny je AUC = 6 mg^min/ml pouze
Úpravy dávkování během léčby nemalobuněčného karcinomu plic:
Abraxane se nemá podávat 1. den cyklu, dokud absolutní počet neutrofilů nedosáhne hodnoty >1 500 buněk/mm3 a počet trombocytů >100 000 buněk/mm3. Pro každou následující týdenní dávku přípravku Abraxane musí mít pacienti absolutní počet neutrofilů >500 buněk/mm3 a počet krevních destiček >50 000 buněk/mm3, jinak je nutné podání dávky pozastavit, dokud se počty těchto krevních elementů nezvýší. Po zvýšení počtu těchto krevních elementů pokračujte v podávání následující týden podle kritérií uvedených v tabulce 4. Snižte následující dávku, pouze pokud jsou splněna kritéria uvedená v tabulce 4.
Tabulka 4: Snížení dávky z důvodu hematologické toxicity u pacientů s nemalobuněčným karcinomem plic
|
Hematologická toxicita |
Výskyt |
Dávka přípravku Abraxane1 (mg/m2) |
Dávka karboplatiny (AUC mg^min/ml)1 |
|
Minimální ANC <500/mm3 s neutropenickou horečkou > 38 °C NEBO Odložení dalšího cyklu kvůli perzistentní neutropenii2 (minimální ANC <1 500/mm3) NEBO Minimální ANC <500/mm3 po dobu > 1 týden |
První |
75 |
4,5 |
|
Druhý |
50 |
3,0 | |
|
Třetí |
Ukončete léčbu | ||
|
Minimální počet krevních destiček <50 000/mm3 |
První |
75 |
4,5 |
|
Druhý |
Ukončete léčbu | ||
11. den 21denního cyklu snižte současně dávku přípravku Abraxane a karboplatiny. 8. nebo 15. den 21denního cyklu snižte dávku přípravku Abraxane; dávku karboplatiny snižte v následujícím cyklu. 2Maximálně za7 dnů po plánované dávce 1. den následujícího cyklu.
Při kožní toxicitě 2. nebo 3. stupně, průjmu 3. stupně nebo mukositidě 3. stupně je třeba přerušit léčbu, dokud se toxicita nevrátí na stupeň < 1, potom znovu zahajte léčbu podle pokynů uvedených v tabulce 5. U periferní neuropatie > 3. stupně je třeba pozastavit léčbu až do návratu na stupeň < 1.
V léčbě je možné pokračovat na další snížené hladině dávek v následujících cyklech podle pokynů uvedených v tabulce 5. U jakékoli jiné nehematologické toxicity 3. nebo 4 stupně je třeba přeušit léčbu, dokud se toxicita nevrátí na stupeň < 2, potom zahajte léčbu podle pokynů uvedených v tabulce 5.
Tabulka 5: Snížení dávky z důvodu nehematologické toxicity u pacientů s nemalobuněčným karcinomem plic____
|
Nehematologická toxicita |
Výskyt |
Dávka přípravku Abraxane1 (mg/m2) |
Dávka karboplatiny (AUC mg^min/ml)1 |
|
Kožní toxicita 2. nebo 3. stupně Průjem 3. stupně Mukositida 3. stupně Periferní neuropatie > 3. stupně Jakákoli j iná nehematologická toxicita 3. nebo 4. stupně |
První |
75 |
4,5 |
|
Druhý |
50 |
3,0 | |
|
Třetí |
Ukončete léčbu | ||
|
Kožní toxicita, průjem nebo mukositida 4. stupně |
První |
Ukončete léčbu | |
11. den 21denního cyklu snižte současně dávku přípravku Abraxane a karboplatiny. 8. nebo 15. den 21denního cyklu snižte dávku přípravku Abraxane; dávku karboplatiny snižte v následujícím cyklu.
Pacienti s poruchou funkce jater
U pacientů s mírnou poruchou funkce jater (celkový bilirubin > 1 až < 1,5 x horní hranice normy [ULN] a aspartátaminotransferáza [AST] < 10 x ULN) není nutná žádná úprava dávkování, bez ohledu na indikaci. Provádějte léčbu stejnými dávkami jako u pacientů s normální funkcí jater.
U pacientů s metastazujícím karcinomem prsu a rovněž u pacientů s nemalobuněčným karcinomem plic se středně těžkou až těžkou poruchou funkce jater (celkový bilirubin > 1,5 až < 5 x ULN a AST < 10 x ULN) se doporučuje snížit dávku o 20 %. Sníženou dávku lze zvýšit na dávku pro pacienty s normální funkcí jater, pokud pacient toleruje léčbu po dobu alespoň dvou cyklů (viz body 4.4 a 5.2).
Pro pacienty s metastazujícím adenokarcinomem pankreatu, kteří mají středně těžkou až těžkou poruchu funkce jater, nejsou dostupné dostatečné údaje, na jejichž základě by bylo možné učinit doporučení ohledně dávkování (viz body 4.4 a 5.2).
Pro pacienty s celkovým bilirubinem > 5 x ULN nebo AST > 10 x ULN nejsou dostupné dostatečné údaje, na jejichž základě by bylo možné učinit doporučení ohledně dávkování, bez ohledu na indikaci (viz body 4.4 a 5.2).
Pacienti s poruchou funkce ledvin
Úprava počáteční dávky přípravku Abraxane není nutná u pacientů s mírnou až středně těžkou poruchou funkce ledvin (odhadovaná clearance kreatininu > 30 až < 90 ml/min). Nejsou dostupné dostatečné údaje pro doporučení úpravy dávkování přípravku Abraxane u pacientů s těžkou poruchou funkce ledvin nebo konečným stadiem renálního selhání (odhadovaná clearance kreatininu < 30 ml/min) (viz bod 5.2).
Starší osoby
Pro pacienty starší 65 let není doporučeno žádné další snížení dávkování kromě toho, které platí pro všechny pacienty.
Z 229 pacientů v randomizované studii, kterým byl podáván přípravek Abraxane v monoterapii k léčbě karcinomu prsu, bylo 13 % ve věku nejméně 65 let a < 2 % bylo věku 75 let a více. U pacientů starších 65 let, kterým byl podáván přípravek Abraxane, se nevyskytovaly žádné výrazně častější toxické účinky. Nicméně, následující analýza provedená u 981 pacientů, kterým byl podáván přípravek Abraxane v monoterapii k léčbě metastatického karcinomu prsu, z nichž 15 % bylo > 65 let a 2 % byla > 75 let, prokázala vyšší incidenci epistaxe, průjmu, dehydratace, únavy a periferních otoků u pacientů > 65 let.
Ze 421 pacientů s karcinomem pankreatu v randomizované studii, kterým byl podáván přípravek Abraxane v kombinaci s gemcitabinem, bylo 41 % ve věku 65 let a více a 10 % ve věku 75 let a více.
U pacientů ve věku 75 let a starších, kterým byl podáván přípravek Abraxane a gemcitabin, byla vyšší incidence závažných nežádoucích účinků a nežádoucích účinků, které vedly k ukončení léčby (viz bod 4.4). Pacienti s karcinomem pankreatu ve věku 75 let a starší by měli být před zvážením léčby důkladně vyšetřeni (viz bod 4.4).
Z 514 pacientů s nemalobuněčným karcinomem plic v randomizované studii, kterým byl podáván přípravek Abraxane v kombinaci s karboplatinou, bylo 31 % ve věku 65 let a více a 3,5 % ve věku 75 let a více. Příhody myelosuprese, příhody periferní neuropatie a artralgie byly častější u pacientů ve věku 65 let a více ve srovnání s pacienty mladšími než 65 let věku. Existují omezené zkušenosti s použitím přípravku Abraxane/karboplatiny u pacientů ve věku 75 let a více.
Farmakokinetické/farmakodynamické modely s využitím údajů od 125 pacientů s pokročilými solidními nádory naznačují, že pacienti > 65 let věku mohou být náchylnější k rozvoji neutropenie během prvního léčebného cyklu.
Pediatrická populace
Bezpečnost a účinnost přípravku Abraxane u dětí a dospívajících ve věku 0-17 let nebyla stanovena. Neexistuje žádné relevantní použití přípravku Abraxane u pediatrické populace v indikaci metastazujícího karcinomu prsu, karcinomu pankreatu nebo nemalobuněčného karcinomu plic.
Způsob podání
Podávejte rekonstituovanou suspenzi přípravku Abraxane nitrožilně pomocí infuzního setu s vestavěným 15 pm filtrem. Po podání se doporučuje propláchnout nitrožilní linku injekčním roztokem chloridu sodného o koncentraci 9 mg/ml (0,9 %), aby bylo zajištěno podání celé dávky.
Návod k rekonstituci tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Laktace (viz bod 4.6).
Pacienti, kteří mají před léčbou počet neutrofilů <1 500 buněk/mm3.
4.4 Zvláštní upozornění a opatření pro použití
Abraxane obsahuje paklitaxel ve formě nanočástic vázaných na albumin. Může mít podstatně odlišné farmakologické vlastnosti v porovnání s dalšími přípravky, které obsahují paklitaxel (viz bod 5.1 a 5.2). Neměl by nahrazovat jiné formy paklitaxelu a ani by jimi neměl být nahrazován.
Hypersenzitivita
Vzácně byl hlášen výskyt těžkých hypersenzitivních reakcí, včetně velmi vzácných anafylaktických reakcí s fatálním průběhem. Jestliže se projeví hypersenzitivní reakce, je nutné neprodleně přerušit podávání přípravku, zahájit symptomatickou léčbu a paklitaxel nesmí být pacientovi znovu podán.
Hematologie
Při léčbě přípravkem Abraxane často dochází k útlumu krvetvorby v kostní dřeni (především k neutropenii). Neutropenie je závislá na dávce a je jí omezena. Během léčby přípravkem Abraxane je nutné často provádět kontrolu krevního obrazu. Další cyklus podávání přípravku Abraxane nelze zahájit, dokud se neutrofilní leukocyty neobnoví na >1 500 buněk/mm3 a trombocyty na >100 000 buněk/mm3 (viz bod 4.2).
Neuropatie
Při léčbě přípravkem Abraxane se často vyskytuje senzorická neuropatie, ale výskyt závažných symptomů je méně častý. Výskyt senzorické neuropatie 1. nebo 2. stupně obvykle nevyžaduje snížení dávkování. Jestliže se při podávání přípravku Abraxane v monoterapii vyvine neuropatie 3. stupně, je nutné léčbu pozastavit, dokud neklesne na stupeň 1 až 2; a pro všechny následné cykly se dále doporučuje pokračovat sníženou dávkou přípravku Abraxane (viz bod 4.2). Jestliže se vyvine neuropatie 3. nebo vyššího stupně při podávání kombinace přípravku Abraxane a gemcitabinu, pozastavte podávání přípravku Abraxane; pokračujte v léčbě gemcitabinem ve stejné dávce. Když periferní neuropatie dosáhne stupně 0 nebo 1, pokračujte v podávání přípravku Abraxane ve snížené dávce (viz bod 4.2). Jestliže se vyvine periferní neuropatie 3. nebo vyššího stupně při podávání kombinace přípravku Abraxane a karboplatiny, je nutné léčbu pozastavit, dokud periferní neuropatie neklesne na stupeň 0 nebo 1, a ve všech následných cyklech se pokračuje sníženou dávkou přípravku Abraxane a karboplatiny (viz bod 4.2).
Sepse
Sepse byla hlášena u 5 % pacientů s neutropenií nebo bez neutropenie, kterým byl podáván přípravek Abraxane v kombinaci s gemcitabinem. Komplikace související se základním onemocněním karcinomem pankreatu, zejména obstrukce žlučovodů nebo přítomnost biliárního stentu, byly identifikovány jako významně se podílející faktory. Jestliže pacient dostane horečku (bez ohledu na
Pneumonitida
Pneumonitida se objevila u 1 % pacientů, kde byl přípravek Abraxane podáván v monoterapii, a u 4 % pacientů, kde byl přípravek Abraxane použit v kombinaci s gemcitabinem. Pečlivě sledujte u všech pacientů výskyt známek a příznaků pneumonitidy. Po vyloučení infekční etiologie a po stanovení diagnózy pneumonitidy natrvalo ukončete léčbu přípravkem Abraxane a gemcitabinem a ihned zahajte vhodnou léčbu a učiňte podpůrná opatření (viz bod 4.2).
Poškození jater
Vzhledem k tomu, že toxicita paklitaxelu může být zvýšena při poškození jater, u pacientů s poškozením jater je třeba opatrnosti při podávání Abraxane. Pacienti s poškozením jater mohou být vystaveni zvýšenému riziku toxicity, zvláště z důvodu myelosuprese; měli by být pozorně sledováni, zda se u nich nevyvine těžká myelosuprese.
Přípravek Abraxane se nedoporučuje u pacientů s celkovým bilirubinem > 5 x ULN nebo AST > 10 x ULN. Kromě toho se přípravek Abraxane nedoporučuje u pacientů s metastazujícím adenokarcinomem pankreatu, kteří mají středně těžkou až těžkou poruchu funkce jater (celkový bilirubin > 1,5 x ULN nebo AST < 10 x ULN) (viz bod 5.2).
Kardiotoxicita
U jedinců, kterým byl podáván Abraxane, byly zaznamenány vzácné případy kongestivního srdečního selhání a dysfunkce levé komory. Většina těchto jedinců byla předtím vystavena léčivým přípravkům s kardiotoxickým účinkem, jako např. antracyklinům, nebo měla v anamnéze srdeční onemocnění. Lékaři by měli pozorně sledovat pacienty v léčbě přípravkem Abraxane z důvodu možného výskytu srdeční příhody.
Metastázy v CNS
Nebyla zjištěna účinnost a bezpečnost přípravku Abraxane u pacientů s metastázami v centrální nervové soustavě (CNS). Systémová chemoterapie se obecně příliš neuplatňuje v léčbě metastáz v CNS.
Gastrointestinální symptomy
Jestliže se po podání přípravku Abraxane dostaví nauzea, zvracení nebo průjem, lze podávat běžně užívaná antiemetika a obstipancia.
Pacienti ve věku 75 let a starší
U pacientů ve věku 75 let a starších nebyly prokázány žádné výhody kombinované léčby Abraxane a gemcitabinu ve srovnání s monoterapií gemcitabinem. U velmi starých osob (> 75 let), kterým byl podáván přípravek Abraxane a gemcitabin, byla vyšší incidence závažných nežádoucích účinků a nežádoucích účinků, které vedly k ukončení léčby. Tyto nežádoucí účinky zahrnovaly hematologickou toxicitu, periferní neuropatii, sníženou chuť k jídlu a dehydrataci. Pacienti s karcinomem pankreatu ve věku 75 let a více by měli být důkladně vyšetřeni z hlediska své schopnosti tolerovat přípravek Abraxane v kombinaci s gemcitabinem, se zvláštním ohledem na stav výkonnosti, další choroby a zvýšené riziko infekcí (viz bod 4.2 a 4.8).
Jiné
Ačkoliv jsou k dispozici pouze omezené údaje, nebyla u pacientů s karcinomem pankreatu s normálními hladinami CA 19-9 před zahájením léčby přípravkem Abraxane a gemcitabinem prokázán jasný prospěch ve smyslu prodlouženého celkového přežití (viz bod 5.1).
Erlotinib nesmí být podáván v kombinaci s přípravkem Abraxane a gemcitabinem (viz bod 4.5).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Metabolismus paklitaxelu je částečně katalyzován izoenzymy CYP2C8 a CYP3A4 cytochromu P450 (viz bod 5.2). Vzhledem k absenci farmakokinetických studií lékových interakcí je proto třeba dbát zvýšené opatrnosti při podávání paklitaxelu současně s přípravky, o nichž je známo, že inhibují CYP2C8 nebo CYP3A4 (např. ketokonazol a jiná imidazolová antimykotika, erytromycin, fluoxetin, gemfibrozil, cimetidin, ritonavir, saquinavir, indinavir a nelfinavir) nebo je indukují (např. rifampicin, karbamazepin, fenytoin, efavirenz, nevirapin).
Paklitaxel a gemcitabin nepodléhají stejnému metabolickému zpracování. Clearance paklitaxelu primárně určuje metabolismus řízený CYP2C8 a CYP3A4, následovaný exkrecí žlučí, zatímco gemcitabin se inaktivuje cytidin-deaminázou, po níž následuje exkrece močí. Farmakokinetické interakce mezi přípravkem Abraxane a gemcitabinem nebyly u lidí hodnoceny.
U pacientů s nemalobuněčným karcinomem plic byla provedena farmakokinetická studie s přípravkem Abraxane a karboplatinou. Nebyly zjištěny žádné klinicky relevantní farmakokinetické interakce mezi přípravkem Abraxane a karboplatinou.
Abraxane je indikován v monoterapii pro léčbu karcinomu prsu, v kombinaci s gemcitabinem pro léčbu karcinomu pankreatu nebo v kombinaci s karboplatinou pro léčbu nemalobuněčného karcinomu plic (viz bod 4.1). Abraxane nesmí být používán v kombinaci s dalšími protirakovinovými přípravky.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Ženy ve fertilním věku by měly během léčby přípravkem Abraxane a ještě l měsíc po ukončení terapie používat účinnou antikoncepci. Mužům léčeným přípravkem Abraxane se doporučuje nepočít dítě během léčby a ne dříve než šest měsíců po ukončení léčby.
O užívání paklitaxelu v těhotenství u žen existují jen velmi omezené údaje. Existuje podezření, že paklitaxel podávaný během těhotenství způsobuje těžké vrozené vady. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3). Abraxane nesmí být užíván v těhotenství a ženami v plodném věku, které nepoužívají účinné antikoncepční metody, s výjimkou případů, kdy klinický stav matky vyžaduje léčbu paklitaxelem.
Kojení
Není známo, zda se paklitaxel vylučuje do lidského mateřského mléka. Vzhledem k možnému výskytu závažných nežádoucích účinků u kojených dětí je Abraxane kontraindikován během kojení. Po dobu trvání léčby musí být kojení přerušeno.
Fertilita
Abraxane způsobil neplodnost u samců potkanů (viz bod 5.3). Muži by se před zahájením léčby měli poradit o uchování spermatu, protože po léčbě přípravkem Abraxane existuje možnost trvalé neplodnosti.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Abraxane má malý nebo mírný vliv na schopnost řídit nebo obsluhovat stroje. Abraxane může vyvolat nežádoucí reakce, například únavu (velmi často) a závratě (často), které mohou ovlivnit schopnost řídit dopravní prostředky a obsluhovat stroje. Pacienty je třeba informovat, aby neřídili dopravní prostředky a neobsluhovali stroje, pokud se cítí unavení nebo pociťují závrať.
Souhrn bezpečnostního profilu
Nejčastější klinicky významné nežádoucí účinky spojené s použitím přípravku Abraxane byly neutropenie, periferní neuropatie, artralgie/myalgie a gastrointestinální poruchy.
Četnost nežádoucích účinků spojených s podáváním přípravku Abraxane je uvedena v tabulce 6 (Abraxane v monoterapii), v tabulce 7 (Abraxane v kombinaci s gemcitabinem) a v tabulce 9 (Abraxane v kombinaci s karboplatinou).
Četnosti jsou definovány jako: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1 000 až <1/100), vzácné (>1/10 000 až <1/1 000), velmi vzácné (<1/10 000). V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
Karcinom prsu (přípravek Abraxane podáván v monoterapii)
Seznam nežádoucích účinků v tabulce
Tabulka 6 uvádí nežádoucí účinky spojené s podáním přípravku Abraxane pacientům ve studiích, v nichž byl Abraxane podáván v monoterapii v jakékoli dávce při jakékoli indikaci (N = 789).
Tabulka 6: Nežádoucí účinky hlášené při podávání přípravku Abraxane v monoterapii v jakýchkoli dávkách v klinických studiích_
|
Infekce a infestace |
Časté: infekce, infekce močových cest, folikulitida, infekce horních cest dýchacích, kandidóza, sinusitida Méně časté: orální kandidóza, nazofaryngitida celulitida, herpes simplex, virová infekce, pneumonie, infekce spojené se zavedením katétru, mykózy, pásový opar, infekce v místě vpichu, sepse2, neutropenická sepse2 |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a p°lypy) |
Méně časté: bolesti související s metastázami, nekróza tumoru |
|
Poruchy krve a lymfatického systému |
Velmi časté: neutropenie, anemie, leukopenie, trombocytopenie, lymfopenie, suprese kostní dřeně Časté: febrilní neutropenie Vzácné: pancytopenie |
|
Poruchy imunitního systému |
Méně časté1: hypersenzitivita Vzácné: závažná hypersenzitivita |
|
Poruchy metabolismu a výživy |
Velmi časté: anorexie Časté: dehydratace, snížená chuť k jídlu, hypokalemie Méně časté: hypofosfatemie, retence tekutin, hypalbuminemie, polydipsie, hyperglykemie, hypokalcemie, hypoglykemie, hyponatremie |
|
Psychiatrické poruchy | |
|
Poruchy nervového systému |
Velmi časté: periferní neuropatie, neuropatie, hypestezie, parestezie Časté: periferní senzorická neuropatie, bolest hlavy, dysgeuzie (poruchy chuti), závratě, periferní motorická neuropatie, ataxie, senzorická porucha, somnolence Méně časté: polyneuropatie, areflexie, dyskinezie, hyporeflexie, neuralgie, |
|
ztráta citlivosti, synkopa, posturální závratě, neuropatická bolest, třes | |
|
Poruchy oka |
Časté: zvýšené slzení, rozmazané vidění, suché oči, keratoconjunctivitis sicca, madaróza Méně časté: podráždění očí, bolest očí, abnormální vidění, snížená ostrost vidění, konjunktivitida, poruchy vidění, svědění očí, keratitida Vzácné: Cystoidní edém makuly2 |
|
Poruchy ucha a labyrintu |
Časté: vertigo Méně časté: otalgie, tinitus |
|
Srdeční poruchy |
Časté: tachykardie, arytmie, supraventrikulární tachykardie Vzácné: bradykardie, zástava srdce, dysfunkce levé komory, kongestivní srdeční selhání, atrioventrikulární blokáda2 |
|
Cévní poruchy |
Časté: zrudnutí, návaly horka, hypertenze, lymfedém Méně časté: hypotenze, studená akra, ortostatická hypotenze Vzácné: trombóza |
|
Respirační, hrudní a mediastinální poruchy |
Časté: intersticiální pneumonitida3, dyspnoe, epistaxe, faryngolaryngální bolest, kašel, rýma, rhinorrhea Méně časté: produktivní kašel, námahová dušnost, kongesce sliznic vedlejších nosních dutin, oslabené dýchání, pleurální výpotek, alergická rýma, chrapot, ucpaný nos, vysychání nosní sliznice, dýchavičnost, plicní embolie, plicní tromboembolie |
|
Gastrointestinální poruchy |
Velmi časté: nauzea, průjem, zvracení, zácpa, stomatitida Časté: bolesti břicha, abdominální distenze, bolesti nadbřišku, dyspepsie, gastroezofageální reflux, hypestezie v ústní dutině Méně časté: dysfagie, flatulence, glosodynie, sucho v ústech, bolesti dásní, řídká stolice, ezofagitida, bolesti v podbřišku, ulcerace úst, bolesti v ústech, rektální krvácení |
|
Poruchy jater a žlučových cest |
Méně časté: hepatomegalie |
|
Poruchy kůže a podkožní tkáně |
Velmi časté: alopecie, vyrážka Časté: porucha nehtů, svědění, suchá kůže, erytém, pigmentace/diskolorace nehtů, hyperpigmentace kůže, onycholýza (uvolnění nehtu z lůžka), změny nehtů Méně časté: bolestivost nehtového lůžka, kopřivka, bolestivost kůže, fotosenzitivní reakce, porucha pigmentace, svědivá vyrážka, porucha kůže, hyperhidróza, onychomadesis (ztráta nehtů), erytematózní vyrážka, celková vyrážka, dermatitida, noční pocení, makulopapulózní vyrážka, vitiligo, hypotrichóza, onemocnění nehtů, celkové svědění, makulární vyrážka, papulózní vyrážka, kožní léze, otoky obličeje Velmi vzácné: Stevensův-Johnsonův syndrom2, toxická epidermální nekrolýza2 |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté: artralgie, myalgie Časté: bolesti v končetinách, bolesti kostí, bolesti zad, svalové křeče, |
|
bolesti končetin Méně časté: bolesti hrudníku, svalová slabost, bolesti šíje, bolesti třísel, svalové spasmy, bolesti svalstva a kostí, bolesti v bocích, nepříjemné pocity v končetinách, svalová slabost | |
|
Poruchy ledvin a močových cest |
Méně časté: dysurie, polakisurie, hematurie, nykturie, polyurie, inkontinence moči |
|
Poruchy reprodukčního systému a prsu |
Méně časté: bolesti prsou |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté: únava, astenie, horečka Časté: periferní otoky, záněty sliznice, bolesti, třesavka, edém, slabost, snížená výkonnost, bolesti na hrudníku, onemocnění podobné chřipce, malátnost, letargie, hyperpyrexie Méně časté: pocit tlaku na hrudi, poruchy chůze, otoky, reakce v místě vpichu Vzácné: extravazace |
|
Vyšetření |
Časté: úbytek tělesné hmotnosti, zvýšení alaninaminotransferázy, zvýšení aspartátaminotransferázy, snížení hematokritu, snížení počtu erytrocytů, zvýšení tělesné teploty, zvýšení gama-glutamyltransferázy, zvýšení alkalické fosfatázy v krvi Méně časté: zvýšený krevní tlak, nárůst hmotnosti, zvýšení laktátdehydrogenázy v krvi, zvýšení kreatininu v krvi, zvýšení krevní glukózy, zvýšená hladina fosforu v krvi, snížená hladina draslíku v krvi, zvýšená hladina bilirubinu |
|
Poranění, otravy a procedurální komplikace |
Méně časté: pohmožděniny Vzácné: návrat postradiačních symptomů (radiation recall phenomenon), radiační pneumonitida |
MedDRA = slovník medicínské terminologie pro regulační činnosti
SMQ = standardizované dotazy MedDRA; SMQ je seskupení několika termínů preferovaných MedDRA k vystižení medicínského pojmu.
1 Četnost hypersenzitivních reakcí je definována na základě jednoznačně určené příčiny ve skupině 789 pacientů.
2 Dle hlášení vycházejících ze sledování přípravku Abraxane po uvedení na trh.
3 Četnost pneumonitidy se počítá na základě sdružených dat od 1 310 pacientů v klinických hodnoceních s přípravkem Abraxane podávaným v monoterapii k léčbě karcinomu prsu a v dalších indikacích zahrnutých MedDRA SMQ pod pojem intersticiální onemocnění plic. Viz bod 4.4.
Popis vybraných nežádoucích účinků
Uvádíme výskyt nejčastějších a klinicky významných nežádoucích účinků u 229 nemocných s metastazujícím karcinomem prsu, kteří byli léčeni přípravkem Abraxane v dávce 260 mg/m2 jednou za tři týdny, v pivotní klinické studii fáze III.
Poruchy krve a lymfatického systému
Nejčastěji pozorovanou významnou hematologickou toxicitou byla neutropenie (hlášena u 79 % pacientů), která byla rychle reversibilní a byla závislá na dávce; leukopenie byla hlášena u 71 % pacientů. Neutropenie 4. stupně (< 500 buněk/mm3) se vyskytla u 9 % pacientů léčených přípravkem Abraxane. Febrilní neutropenie se vyskytla u 4 pacientů léčených přípravkem Abraxane. Anemie (Hb <10 g/dl) byla pozorována u 46 % pacientů léčených přípravkem Abraxane a ve třech případech byla závažná (Hb <8 g/dl). Lymfopenie byla pozorována u 45 % pacientů.
Poruchy nervového systému
U nemocných léčených přípravkem Abraxane byla četnost a závažnost neurotoxicity obvykle závislá na dávce. Periferní neuropatie (většinou senzorická neuropatie 1. nebo 2. stupně) byla pozorována u 68 % nemocných léčených přípravkem Abraxane, z toho 10 % byla 3. stupně, žádné případy 4. stupně.
Gastrointestinální poruchy
U 29 % nemocných se vyskytla nauzea, u 25 % průjem.
Poruchy kůže a podkožní tkáně
U > 80 % nemocných léčených přípravkem Abraxane byla pozorována alopecie. Většina případů alopecie se vyskytla do jednoho měsíce po zahájení léčby přípravkem Abraxane. U většiny pacientů, u nichž se vyskytne alopecie, se očekává výrazná ztráta vlasů > 50 %.
Poruchy svalové a kosterní soustavy a pojivové tkáně
Artralgie se vyskytla u 32 % nemocných léčených přípravkem Abraxane a v 6 % případů byla závažná. Myalgie se vyskytla u 24 % nemocných léčených přípravkem Abraxane a v 7 % případů byla závažná. Symptomy byly obvykle přechodné, typicky se vyskytovaly tři dny po podání přípravku Abraxane a do týdne vymizely.
Celkové poruchy a reakce v místě aplikace Astenie/únava byla hlášena u 40 % nemocných.
Karcinom pankreatu (přípravek Abraxane podávaný v kombinaci s gemcitabinem)
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky byly hodnoceny u 421 pacientů léčených přípravkem Abraxane v kombinaci s gemcitabinem a u 402 pacientů léčených gemcitabinem v monoterapii, jako systémová léčba první linie metastazujícího karcinomu pankreatu, v randomizovaném, kontrolovaném, otevřeném hodnocení fáza III. V tabulce 7 jsou uvedeny nežádoucí účinky hodnocené u pacientů s karcinomem pankreatu, kteří byli léčeni přípravkem Abraxane v kombinaci s gemcitabinem.
Tabulka 7: Nežádoucí účinky hlášené při léčbě přípravkem Abraxane v kombinaci
|
s gemcitabinem (N = 421 | |
|
Infekce a infestace |
Časté: sepse, pneumonie, orální kandidóza |
|
Poruchy krve a lymfatického systému |
Velmi časté: neutropenie, anemie, trombocytopenie Časté: pancytopenie Méně časté: trombotická trombocytopenická purpura |
|
Poruchy metabolismu a výživy |
Velmi časté: dehydratace, snížená chuť k jídlu, hypokalemie |
|
Psychiatrické poruchy | |
|
Poruchy nervového systému |
Velmi časté: periferní neuropatie1, dysgeuzie, bolest hlavy, závratě Méně časté: paralýza VII. hlavového nervu |
|
Poruchy oka |
Časté: zvýšené slzení Méně časté: cystoidní makulární edém |
|
Srdeční poruchy |
Časté: kongestivní srdeční selhání, tachykardie |
|
Cévní poruchy |
Časté: hypotenze, hypertenze |
|
s gemcitabinem (N = 421 | |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté: dyspnoe, epistaxe, kašel Časté: pneumonitida2, ucpaný nos Méně časté: sucho v krku, vysychání nosní sliznice |
|
Gastrointestinální poruchy |
Velmi časté: nauzea, průjem, zvracení, zácpa, bolesti břicha, bolesti nadbřišku Časté: stomatitida, obstrukce střeva, kolitida, sucho v ústech |
|
Poruchy jater a žlučových cest |
Časté: cholangitida |
|
Poruchy kůže a podkožní tkáně |
Velmi časté: alopecie, vyrážka Časté: svědění, suchá kůže, porucha nehtů, zrudnutí |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté: bolesti v končetinách, artralgie, myalgie Časté: svalová slabost, bolesti kostí |
|
Poruchy ledvin a močových cest |
Časté: akutní selhání ledvin Méně časté: hemolyticko-uremický syndrom |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté: únava, periferní otoky, horečka, astenie, zimnice Časté: reakce v místě podání infuze |
|
Vyšetření |
Velmi časté: úbytek tělesné hmotnosti, zvýšení alaninaminotransferázy Časté: zvýšení aspartátaminotransferázy, zvýšení bilirubinu v krvi, zvýšení kreatininu v krvi |
MedDRA = slovník medicínské terminologie pro regulační činnosti; SMQ = standardizované dotazy MedDRA (seskupení několika termínů preferovaných MedDRA k vystižení medicínského pojmu).
1 Periferní neuropatie hodnocená za použití SMQ (široký rozsah).
2 Pneumonitida se hodnotí za použití SMQ pro intersticiální plicní onemocnění (široký rozsah).
V tomto randomizovaném, kontrolovaném, otevřeném hodnocení fáze III byly nežádoucí účinky, které vedly k úmrtí do 30 dnů od poslední dávky sledovaného léku, hlášeny u 4 % pacientů, kterým byl podáván přípravek Abraxane v kombinaci s gemcitabinem, a u 4 % pacientů, kteří byli léčeni gemcitabinem v monoterapii.
Popis vybraných nežádoucích účinků
Uvádíme výskyt nejčastějších a nejdůležitějších nežádoucích účinků u 421 nemocných s metastazujícím karcinomem pankreatu, kteří byli léčeni přípravkem Abraxane v dávce 125 mg/m2 v kombinaci s gemcitabinem v dávce 1 000 mg/m2 podávanými 1., 8. a 15. den každého 28denního cyklu v klinické studii fáze III.
Poruchy krve a lymfatického systému
V tabulce 8 je uvedena četnost a závažnost laboratorně zjištěných hematologických abnormalit u pacientů léčených přípravkem Abraxane v kombinaci s gemcitabinem nebo u pacientů léčených gemcitabinem.
Tabulka 8: Laboratorně zjištěné hematologické abnormality v hodnocení u pacientů s karcinomem pankreatu___
|
Abraxane (125 mg/m2)/ gemcitabin |
Gemcitabin | |||
|
Stupeň 1-4 (%) |
Stupeň 3-4 (%) |
Stupeň 1-4 (%) |
Stupeň 3-4 (%) | |
|
Anemieab |
97 |
13 |
96 |
12 |
|
Neutropenie ab |
73 |
38 |
58 |
27 |
|
Trombocytopeniebc |
74 |
13 |
70 |
9 |
a 405 pacientů hodnocených ve skupině léčené přípravky Abraxane/gemcitabin b 388 pacientů hodnocených ve skupině léčené gemcitabinem c 404 pacientů hodnocených ve skupině léčené přípravky Abraxane/gemcitabin
Periferní neuropatie
U pacientů léčených přípravkem Abraxane v kombinaci s gemcitabinem byl medián doby do prvního výskytu periferní neuropatie 3. stupně 140 dnů. Medián doby do zlepšení alespoň o 1 stupeň byl 21 dnů a medián doby do zlepšení z periferní neuropatie 3. stupně na stupeň 0 nebo 1 byl 29 dnů.
Z pacientů, u kterých byla léčba z důvodu periferní neuropatie přerušena, se bylo schopno 44 %
(31/70 pacientů) vrátit k léčbě přípravkem Abraxane ve snížené dávce. Žádný z pacientů, kteří byli léčeni přípravkem Abraxane v kombinaci s gemcitabinem, neměl periferní neuropatii 4. stupně.
Sepse
Sepse byla hlášena u 5 % pacientů s neutropenií nebo bez neutropenie, kterým byl v průběhu klinického hodnocení léčby karcinomu pankreatu podáván přípravek Abraxane v kombinaci s gemcitabinem. Jako významné přispívající faktory byly identifikovány komplikace související se základním onemocněním karcinomem pankreatu, zejména obstrukce žlučovodů nebo přítomnost biliárního stentu. Jestliže pacient dostane horečku (bez ohledu na počet neutrofilů), zahajte léčbu širokospektrálními antibiotiky. Při febrilní neutropenii pozastavte podávání přípravku Abraxane a gemcitabinu, dokud horečka neustoupí a absolutní počet neutrofilů nedosáhne >1 500 buněk/mm3, poté pokračujte v podávání snížených dávek (viz bod 4.2).
Pneumonitida
Pneumonitida byla hlášena u 4 % pacientů při použití přípravku Abraxane v kombinaci s gemcitabinem. Ze 17 hlášených případů pneumonitidy u pacientů léčených přípravkem Abraxane v kombinaci s gemcitabinem měly 2 fatální následky. Pečlivě sledujte u všech pacientů výskyt známek a příznaků pneumonitidy. Po vyloučení infekční etiologie a po stanovení diagnózy pneumonitidy natrvalo ukončete léčbu přípravkem Abraxane a gemcitabinem a ihned zahajte vhodnou léčbu a učiňte podpůrná opatření (viz bod 4.2).
Nemalobuněčný karcinom plic (přípravek Abraxane podávaný v kombinaci s karboplatinou)
Seznam nežádoucích účinků v tabulce
V tabulce 9 jsou uvedeny nežádoucí účinky spojené s podáváním přípravku Abraxane v kombinaci s karboplatinou.
|
Infekce a infestace |
Časté: pneumonie, bronchitida, infekce horních cest dýchacích, infekce močových cest Méně časté: sepse, orální kandidóza |
|
Poruchy krve a lymfatického systému1 |
Velmi časté: neutropenie1, trombocytopenie1, anemie1, leukopenie1 Časté: febrilní neutropenie, lymfopenie Méně časté: pancytopenie |
|
Poruchy imunitního systému |
Méně časté: léková hypersenzitivita, hypersenzitivita |
|
Poruchy metabolismu a výživy |
Velmi časté: snížená chuť k jídlu Časté: dehydratace |
|
Psychiatrické poruchy |
Časté: nespavost |
|
Poruchy nervového systému |
Velmi časté: periferní neuropatie2 Časté: dysgeuzie, bolest hlavy, závratě |
|
Poruchy oka |
Časté: rozmazané vidění |
|
Cévní poruchy |
Časté: hypotenze, hypertenze Méně časté: zarudnutí |
|
Respirační, hrudní a mediastinální poruchy |
Velmi časté: dyspnoe Časté: hemoptýza, epistaxe, kašel Méně časté: pneumonitida3 |
|
Gastrointestinální poruchy |
Velmi časté: průjem, zvracení, nauzea, zácpa Časté: stomatitida, dyspepsie, bolesti břicha, dysfágie |
|
Poruchy jater a žlučových cest |
Časté: hyperbilirubinemie |
|
Poruchy kůže a podkožní tkáně |
Velmi časté: vyrážka, alopecie Časté: svědění, porucha nehtů Méně časté: exfoliace kůže, alergická dermatitida, kopřivka |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
Velmi časté: artralgie, myalgie Časté: bolesti zad, bolesti v končetinách, muskuloskeletální bolest |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté: únava, astenie, periferní otoky Časté: horečka, bolest na hrudi Méně časté: zaněty sliznice, extravazace v místě infuze, zánět v místě infuze, vyrážka v místě infuze |
|
Vyšetření |
Časté: zvýšení alaninaminotransferázy, zvýšení aspartátaminotransferázy, zvýšení alkalické fosfatázy v krvi, úbytek tělesné hmotnosti |
MedDRA = slovník medicínské terminologie pro regulační činnosti: SMQ = standardizované dotazy MedDRA
1 Vychází z laboratorních hodnocení: maximální stupeň myeolosuprese (léčená populace)
2 Periferní neuropatie se hodnotí za použití SMQ pro neuropatii (široký rozsah)
3 Pneumonitida se hodnotí za použití SMQ pro intersticiální plicní onemocnění (široký rozsah)
U pacientů s nemalobuněčným karcinomem plic léčených přípravkem Abraxane a karboplatinou byl medián doby do prvního výskytu periferní neuropatie spojené s léčbou 3. stupně 121 dnů a medián doby do návratu periferní neuropatie související s léčbou 3. stupně na stupeň 1 byl 38 dnů. U žádného pacienta léčeného přípravkem Abraxane a karboplatinou nebyla zaznamenána periferní neuropatie 4. stupně.
Ve větvi s přípravkem Abraxane byly anemie a trombocytopenie hlášeny častěji než ve větvi s přípravkem Taxol (54 % versus 28 %, resp. 45 % versus 27 %).
Toxicita taxanu hlášená pacienty se hodnotila za použití 4 podstupnic funkčního hodnocení při léčbě rakoviny (FACT) - dotazníku pro taxan. Při použití analýzy opakovaného měření byla kombinace přípravku Abraxane a karboplatiny favorizována ve 3 ze 4 podstupnic (periferní neuropatie, bolest rukou/nohou a sluch) (p < 0,002). U poslední podskupiny (otok) nebyl mezi léčebnými větvemi zjištěn rozdíl.
Postmarketingová zkušenost
Během postmarketingového sledování přípravku Abraxane byly pozorovány obrny hlavových nervů, paréza hlasivek a vzácně byly hlášeny závažné hypersenzitivní reakce.
Ve vzácných případech byly v průběhu léčby přípravkem Abraxane hlášeny případy snížené zrakové ostrosti v důsledku cystoidního edému makuly. V případě diagnózy cystoidního edému makuly musí být léčba přípravkem Abraxane přerušena.
Jako součást dlouhodobého sledování přípravku Abraxane byly u některých pacientů dříve léčených kapecitabinem hlášeny případy palmar-plantární erytrodysestezie. Vzhledem k tomu, že tyto příhody byly hlášeny dobrovolně během klinické praxe, nelze učinit pravdivé odhady o jejich četnosti a nebyl zjištěn jejich kauzální vztah.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Při předávkování paklitaxelem není známo žádné antidotum. V případě předávkování musí být pacient pozorně sledován. Léčbu je nutno zaměřit na hlavní očekávané toxické účinky, což je suprese kostní dřeně, mukozitida a periferní neuropatie.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Cytostatika, rostlinné alkaloidy a jiná přírodní léčiva, taxany, ATC kód: L01CD01
Mechanismus účinku
Paklitaxel je antimikrotubulová látka, která podporuje shlukování mikrotubulů z tubulinových dimerů a stabilizuje mikrotubuly tím, že brání depolymerizaci. Tato stabilita vede k inhibici normální dynamické reorganizace mikrotubulové sítě, která je nezbytná pro vitální interfázové a mitotické buněčné funkce. Paklitaxel navíc indukuje abnormální svazky mikrotubulů v průběhu celého buněčného cyklu a četné hvězdice mikrotubulů během mitózy.
Abraxane obsahuje nanočástice o velikosti přibližně 130 nm, složené z lidského sérového albuminu a paklitaxelu, ve kterých je paklitaxel přítomen v nekrystalické, tj. amorfní formě. Po intravenózním podání dochází k rozpadu nanočástic na rozpustné komplexy paklitaxelu vázaného na albumin o velikosti přibližně 10 nm. O albuminu je známo, že zprostředkovává endotelovou transcytózu složek plasmy kaveolami, a studie in vitro prokázaly, že přítomnost albuminu v přípravku Abraxane zvyšuje transport paklitaxelu skrz endotelové buňky. Předpokládá se, že tento zvýšený kaveolární transport skrze endotel je zprostředkován receptorem albuminu gp-60 a že v oblasti tumoru je zvýšená akumulace paklitaxelu zprostředkována proteinem SPARC (Secreted Protein Acidic Rich in Cysteine), který váže albumin.
Klinická účinnost a bezpečnost
Karcinom _ prsu
Pro podporu využití přípravku Abraxane při metastazujícím karcinomu prsu jsou k dispozici údaje ze skupiny 106 nemocných, nashromážděné ze dvou otevřených studií s jednou léčebnou větví, a ze skupiny 454 nemocných léčených v randomizované srovnávací studii fáze III. Tyto informace jsou uvedeny níže.
Otevřené studie s jednou léčebnou větví
V jedné studii byl Abraxane podáván ve formě 30 minutové infuze v dávce 175 mg/m2 43 pacientům s metastazujícím karcinomem prsu. Druhá studie používala dávku 300 mg/m2 ve formě 30 minutové infuze podávané 63 nemocným s metastazujícím karcinomem prsu. Pacienti byli léčeni bez předchozí léčby steroidy nebo plánované podpory G-CSF. Cykly byly aplikovány v třítýdenních intervalech. Léčebná odpověď u všech nemocných činila 39,5 % (95% CI: 24,9 % - 54,2 %) a 47,6 %
(95% CI: 35,3 % - 60,0 %). Medián doby do progrese onemocnění byl 5,3 měsíce (175 mg/m2;
95% CI: 4,6 - 6,2 měsíce) a 6,1 měsíce (300 mg/m2; 95% CI: 4,2 - 9,8 měsíce).
Randomizovaná srovnávací studie
Tato multicentrická studie probíhala u nemocných s metastazujícím karcinomem prsu, léčených každé 3 týdny monoterapií paklitaxelem. Paklitaxel byl podáván v rozpustné formě v dávce 175 mg/m2 jako tříhodinová infuze s premedikací pro prevenci hypersenzitivity (N = 225), nebo jako přípravek Abraxane v dávce 260 mg/m2 podávaný jako 30minutová infuze bez premedikace (N = 229).
Šedesát čtyři procent nemocných mělo při zařazení do studie zhoršený stav klinické kondice (ECOG 1 nebo 2); 79 % mělo viscerální metastázy a 76 % mělo metastázy na více než 3 místech. Čtrnáct procent nemocných neprodělalo dřívější chemoterapii; 27 % prodělalo chemoterapii pouze jako adjuvantní léčbu, 40 % pouze pro metastazující onemocnění a 19 % pro metastazující onemocnění i jako adjuvantní léčbu. Padesát devět procent nemocných dostávalo hodnocený léčivý přípravek jako terapii druhé nebo vyšší než druhé linie. Sedmdesát sedm procent nemocných bylo dříve vystaveno působení antracyklinů.
Výsledky celkového podílu léčebné odpovědi a doby do progrese onemocnění, přežití bez progrese a přežití v závislosti na tom, zda šlo o terapii vyšší než první linie, jsou uvedeny níže.
Tabulka 10: Výsledky celkové léčebné odpovědi, mediánu doby do progrese choroby a přežití bez progrese podle hodnocení výzkumného pracovníka
|
Parametry účinnosti |
Abraxane (260 mg/m2) |
Paklitaxel v rozpustné formě (175 mg/m2) |
p-hodnota |
|
Podíl léčebné odpovědi [95% CI] (%) | |||
|
>Terapie první linie |
26,5 [18,98; 34,05] (n = 132) |
13,2 [7,54; 18,93] (n = 136) |
0,006a |
|
*Medián doby do progrese onemocnění [95% CI] (týdny) | |||
|
>Terapie první linie |
20,9 [15,7; 25,9] (n = 131) |
16,1 [15,0; 19,3] (n = 135) |
0,011b |
|
*Medián přežití bez progrese onemocnění [95% CI] (týdny) | |||
|
>Terapie první linie |
20,6 [15,6; 25,9] (n = 131) |
16,1 [15,0; 18,3] (n = 135) |
0,010b |
|
*Přežití [95% CI] (týdny) | |||
|
>Terapie první linie |
56,4 [45,1; 76,9] (n = 131) |
46,7 [39,0; 55,3] (n = 136) |
0,020b |
Tyto údaje jsou založeny na zprávě o klinické studii: CA012-0, dodatek z 23. března 2005 a Chi-kvadrát test b Log-rank test
U 229 pacientů léčených přípravkem Abraxane v randomizovaném kontrolovaném klinickém hodnoceni byla posuzována bezpečnost. Neurotoxicita paklitaxelu byla posuzována na základě zlepšení o jeden stupeň kdykoli během léčby u pacientů se 3. stupněm periferní neuropatie. V důsledku kumulativní toxicity přípravku Abraxane nebyl hodnocen přirozený průběh periferní neuropatie a návrat k počátečnímu stavu po více než 6 cyklech léčby a zůstává neznámý.
Karcinom pankreatu
Byla provedena multicentrická, mezinárodní, randomizovaná, otevřená studie s 861 pacienty ke srovnání léčby přípravkem Abraxane/gemcitabinem a monoterapie gemcitabinem jako léčby první linie u pacientů s metastazujícím karcinomem pankreatu. Přípravek Abraxane byl podáván pacientům (N = 431) nitrožilní infuzí po dobu 30-40 minut v dávce 125 mg/m2, potom následovalo podání gemcitabinu nitrožilní infuzí po dobu 30-40 minut v dávce 1 000 mg/m2. Oba léky se podávaly 1., 8. a 15. den každého 28denního cyklu. Ve srovnávací léčebné větvi byl gemcitabin v monoterapii podáván pacientům (N = 430) podle doporučeného dávkování a režimu. Léčba se prováděla do progrese onemocnění nebo do rozvoje nepřijatelné toxicity. Ze 431 pacientů s karcinomem pankreatu, kteří byli randomizováni do větve s přípravkem Abraxane v kombinaci s gemcitabinem, byla většina (93 %) běloši, 4 % byli černoši a 2 % byli Asiaté. 16 % mělo stav výkonnosti podle Karnofského (Karnofsky performance status, KPS) 100; 42 % mělo KPS 90; 35 % mělo KPS 80; 7 % mělo KPS 70 a <1 % pacientů mělo KPS nižší než 70. Pacienti s vysokým kardiovaskulárním rizikem, anamnézou periferního arteriálního onemocnění a/nebo poruch pojivové tkáně a/nebo intersticiální plicní nemoci byli ze studie vyřazeni.
Medián doby léčby pacientů byl 3,9 měsíců ve větvi s přípravkem Abraxane/gemcitabin a 2,8 měsíců ve větvi s gemcitabinem. 32 % pacientů ve větvi s přípravkem Abraxane/gemcitabin bylo léčeno 6 nebo více měsíců oproti 15 % pacientů ve větvi s gemcitabinem. V léčené populaci byl medián relativní intenzity dávky gemcitabinu 75 % ve větvi s přípravkem Abraxane/gemcitabin a 85 % ve větvi s gemcitabinem. Medián relativní intenzity dávky přípravku Abraxane byl 81 %. Vyšší medián
Primárním cílovým parametrem účinnosti bylo celkové přežití (overall survival, OS). Klíčovými sekundárními cílovými parametry bylo přežití bez progrese (progression-free survival, PFS) a celková míra odpovědi (overall response rate, ORR); oba sekundární cílové parametry byly posouzeny nezávislým, centrálním, zaslepeným radiologickým hodnocením za použití kritérií RECIST (verze 1.0).
Tabulka 11: Výsledky účinnosti z randomizované studie u pacientů s karcinomem pankreatu (populace intent-to-treat) __
|
Abraxane(125 mg/m2)/gemcitabin (N=431) |
Gemcitabin (N=430) | |
|
Celkové přežití | ||
|
Počet úmrtí (%) |
333 (77) |
359 (83) |
|
Medián celkového přežití, měsíce (95% CI) |
8,5 (7,89; 9,53) |
6,7 (6,01; 7,23) |
|
HRa+g/g (95% CI)a |
0,72 (0,617; 0,835) | |
|
p-hodnotab |
<0,0001 | |
|
Míra přežití % (95% CI) | ||
|
v 1. roce |
35 % (29,7; 39,5) |
22 % (18,1; 26,7) |
|
ve 2. roce |
9 % (6,2; 13,1) |
4 % (2,3; 7,2) |
|
75. percentil celkového přežití (měsíce) |
14,8 |
11,4 |
|
Přežití bez progrese | ||
|
Úmrtí nebo progrese, n (%) |
277 (64) |
265 (62) |
|
Medián přežití bez progrese, měsíce (95% CI) |
5,5 (4,47; 5,95) |
3,7 (3,61; 4,04) |
|
HRa+g/g (95% CI)a |
0,69 (0,581 |
; 0,821) |
|
p-hodnotab |
<0,0001 | |
|
Celková míra odpovědi | ||
|
Potvrzená úplná nebo částečná celková odpověď, n (%) |
99 (23) |
31 (7) |
|
95% CI |
19,1; 27,2 |
5,0; 10,1 |
|
pa+g/pg (95% CI) |
3,19 (2,178; 4,662) | |
|
p-hodnota (chí-kvadrát test) |
<0,0001 | |
CI = interval spolehlivosti, HRA+G/G = poměr rizik přípravku Abraxane+gemcitabinu/gemcitabinu, pA+G/pG = poměr míry odpovědi u přípravku Abraxane+gemcitabinu/gemcitabinu a stratifikovaný Coxův model proporcionálních rizik
b stratifikovaný log-rank test, stratifikovaný podle geografických regionů (severní Amerika nebo další), KPS (70 až 80 versus 90 až 100) a přítomnost metastáz v játrech (ano nebo ne).
Bylo zjištěno statisticky významné zlepšení OS u pacientů léčených přípravkem Abraxane/gemcitabin oproti samotnému gemcitabinu s 1,8měsíčním zvýšením mediánu OS, 28% celkovým snížením rizika náhlé smrti, 59% zlepšení v 1roční míře přežití a 125% zlepšení ve 2leté míře přežití.
Obrázek 1: Kaplan-Meierova křivka celkového přežití (populace intent-to-treat)
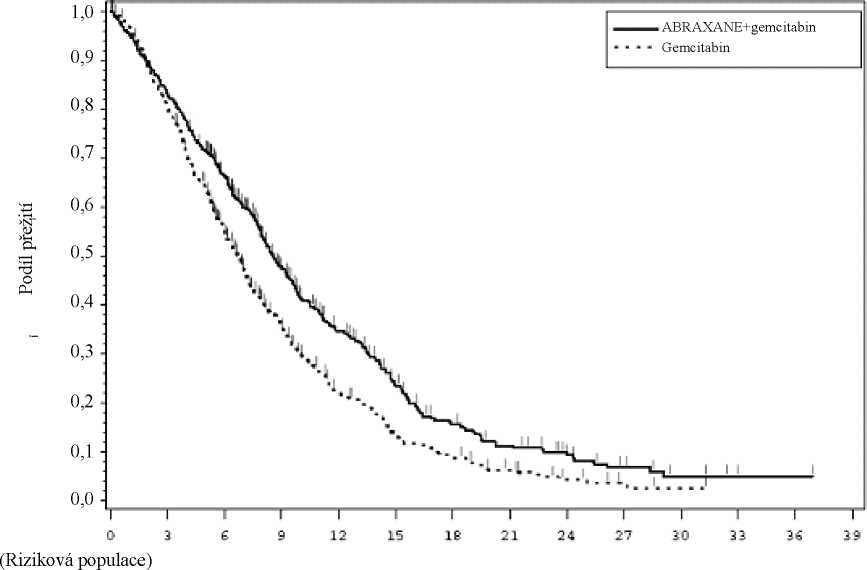
ABX/GEM :
GEM : ■■■■'
257 203
2JO 220
10? 12 4
10 0
es
Í7
40
10
20
15
10
7
Čas (měsíce)
Léčebné účinky na OS favorizovaly větev s přípravkem Abraxane/gemcitabin napříč většinou dříve specifikovaných podskupin (zahrnujících pohlaví, KPS, geografický region, primární lokalizaci karcinomu pankreatu, rozsah nádoru (stage) při diagnóze, přítomnost metastáz v játrech, přítomnost peritoneální karcinomatózy, provedenou Whippleovu operaci, přítomnost biliárního stentu na začátku léčby, přítomnost metastáz v plicích a počet míst metastáz). U pacientů ve věku >75 let byl ve větvi s přípravkem Abraxane/gemcitabin a ve větvi s gemcitabinem poměr rizik přežití 1,08 (95% CI 0,653; 1,797). U pacientů s normálními hladinami CA 19-9 na začátku léčby byl poměr rizik přežití 1,07 (95% CI 0,692; 1,661).
U pacientů léčených přípravkem Abraxane/gemcitabin bylo zaznamenáno statisticky významné zlepšení PFS oproti pacientům léčeným samotným gemcitabinem, a to zvýšení mediánu PFS o 1,8 měsíců.
Nemalobuněčný karcinom _plic
U 1 052 pacientů s nemalobuněčným karcinomem plic stupně IIIB/IV, kteří do té doby neabsolvovali chemoterapii, byla provedena multicentrická, randomizovaná, otevřená studie. Ve studii se porovnával přípravek Abraxane v kombinaci s karboplatinou oproti paklitaxelu v rozpustné formě v kombinaci s karboplatinou jako léčba první linie u pacientů s pokročilým nemalobuněčným karcinomem plic.
Přes 99 % pacientů mělo stav klinické kondice ECOG (Eastern Cooperative Oncology Group)
0 nebo 1. Pacienti s dříve existující neuropatií >2. stupně nebo se závažnými zdravotními rizikovými faktory, týkajícími se jakéhokoli hlavního orgánového systému, byli vyloučeni. Přípravek Abraxane byl podáván pacientům (N = 521) nitrožilní infuzí po dobu 30 minut v dávce 100 mg/m2 1., 8. a 15. den každého 21denního cyklu bez jakékoli premedikace steroidy a bez profylaxe faktorem stimulujícím kolonie granulocytů. Karboplatina v dávce AUC = 6 mg^min/ml byla podávána nitrožilně pouze 1. den každého 21denního cyklu, přičemž podávání bylo zahájeno ihned po dokončení podání přípravku Abraxane. Paklitaxel v rozpustné formě byl podáván pacientům (N = 531) v dávce 200 mg/m2 nitrožilní infuzí po dobu 3 hodin se standardní premedikací, ihned následovalo nitrožilní podání karboplatiny v dávce AUC = 6 mg^min/ml. Každý lék byl podán 1. den každého 21denního
Primárním cílovým parametrem účinnosti byla celková míra odpovědi, definovaná jako procento pacientů, u kterých bylo dosaženo objektivně potvrzené úplné odpovědi nebo částečné odpovědi, založená na nezávislém, centrálním, zaslepeném, radiologickém hodnocení programem RECIST (verzí 1.0). U pacientů ve větvi s přípravkem Abraxane/karboplatinou byla zaznamenána významně vyšší celková míra odpovědi ve srovnání s pacienty v kontrolní větvi: 33 % versus 25 %, p = 0,005 (tabulka 12). Byl nalezen významný rozdíl v celkové míře odpovědi u větve s přípravkem Abraxane/karboplatinou v porovnání s kontrolní větví u pacientů s nemalobuněčným karcinomem plic skvamózní histologie (N = 450, 41 % vs. 24 %, p < 0,001), tento rozdíl se však neprojevil jako rozdíl v kategorii PFS ani OS. V rámci léčebných skupin nebyl rozdíl v ORR u pacientů s neskvamózní histologií (N=602, 26 % vs 25 %, p=0,808).
Tabulka 12: Celková míra odpovědi v randomizované studii nemalobuněčného karcinomu plic (populace intent-to-treat)___
|
Parametr účinnosti |
Přípravek Abraxane (100 mg/m2/týden) + karboplatina (N=521) |
Paklitaxel v rozpustné formě (200 mg/m2 každé 3 týdny) + karboplatina (N=531) |
|
Celková míra odpovědi (nezávislé hodnocení) | ||
|
Potvrzená úplná nebo částečná celková odpověď, n (%) |
170 (33 %) |
132 (25 %) |
|
95% CI (%) |
28,6; 36,7 |
21,2; 28,5 |
|
pA/pT (95,1% CI) |
1,313 (1,082; 1,593) | |
|
p-hodnotaa |
0,005 | |
CI = interval spolehlivosti; HRA/T = poměr rizik přípravku Abraxane/karboplatiny a paklitaxelu v rozpustné formě / karboplatiny; pA/pT = poměr míry odpovědi přípravku Abraxane/karboplatiny a paklitaxelu v rozpustné formě / karboplatiny a p-hodnota vychází z chí-kvadrát testu.
Mezi oběma léčebnými větvemi nebyl nalezen statisticky významný rozdíl v přežití bez progrese (zaslepeným hodnocením radiologa) a celkovém přežití. Analýza non-inferiority byla provedena pro PFS a OS s předem stanovenou hranicí non-inferiority 15 %. Kritérium pro non-inferioritu bylo splněno u PFS i OS s horní hranicí 95% intervalu spolehlivosti pro související poměry rizik s hodnotami nižšími než 1,176 (tabulka 13).
Tabulka 13: Analýzy non-inferiority týkající se přežití bez progrese a celkového přežití v randomizované studii nemalobuněčného karcinomu (populace intent-to-treat)_
|
Parametr účinnosti |
Přípravek Abraxane (100 mg/m2/týden) + karboplatina (N=521) |
Paklitaxel v rozpustné formě (200 mg/m2 každé 3 týdny) + karboplatina (N=531) |
|
Přežití bez progresea (nezávislé hodnocení) | ||
|
Úmrtí nebo progrese, n (%) |
429 (82 %) |
442 (83 %) |
|
Medián PFS (95% CI) (měsíce) |
6,8 (5,7; 7,7) |
6,5 (5,7; 6,9) |
|
HRa/t (95% CI) |
0,949 (0,830; 1,086) | |
|
Celkové přežití | ||
|
Počet úmrtí, n (%) |
360 (69 %) |
384 (72 %) |
|
Medián OS (95% CI) (měsíce) |
12,1 (10,8; 12,9) |
11,2 (10,3; 12,6) |
|
HRa/t (95,1% CI) |
0,922 (0,797; 1,066) | |
CI = interval spolehlivosti; HRA/T = poměr rizik přípravku Abraxane/karboplatiny a paklitaxelu v rozpustné formě / karboplatiny; pA/pT = poměr míry odpovědi přípravku Abraxane/karboplatiny a paklitaxelu v rozpustné formě / karboplatiny
a Podle metodologických kritérií EMA pro cílový parametr PFS nebyla chybějící pozorování nebo zahájení následné nové terapie použita pro cenzurování.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Abraxane u všech podskupin pediatrické populace, které podstupují léčbu metastazujícího karcinomu prsu, karcinomu pankreatu a nemalobuněčného karcinomu plic (informace o použití u dětí viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika celkově podaného paklitaxelu v infuzích v trvání 30 a 180 minut, přípravku Abraxane v dávkování 80 až 375 mg/m2, byla stanovena v klinických studiích. Expozice paklitaxelem (křivka AUC) stoupala lineárně od 2 653 do 16 736 ng.hod/ml při dávkování od 80 do 300 mg/m2.
Ve studii nemocných s pokročilými solidními tumory byly srovnávány farmakokinetické charakteristiky paklitaxelu po intravenózní aplikaci přípravku Abraxane v dávce 260 mg/m2 po dobu 30 minut s charakteristikami paklitaxelu v rozpustné formě v dávce 175 mg/m2, podávaného injekcí po dobu 3 hodin. Z non kompartmentové farmakokinetické analýzy vyplývá, že plazmatická clearance paklitaxelu s přípravkem Abraxane byla větší (43 %) než po injekci paklitaxelu na bázi rozpouštědla a také zde byl vyšší distribuční objem (53 %). Konečné poločasy se nelišily.
Ve studii s opakovaným dávkováním provedené na 12 pacientech, kterým byl intravenózně podáván přípravek Abraxane v dávce 260 mg/m2, byla variabilita mezi pacienty v AUC 19 % (rozmezí =
3,21 % - 27,70 %). Nebyla prokázána akumulace paklitaxelu při vícenásobných léčebných kúrách.
Distribuce
Po podání přípravku Abraxane pacientům se solidními tumory se paklitaxel rovnoměrně distribuuje do krevních buněk a plazmy a ve vysoké míře se váže na bílkoviny krevní plazmy (94 %).
Vazba paklitaxelu na bílkoviny po podání přípravku Abraxane, byla stanovena ultrafiltrací v rámci srovnávací studie u pacienta. Frakce volného paklitaxelu byla signifikantně vyšší u přípravku Abraxane (6,2 %) než u paklitaxelu založeném na bázi rozpouštědla (2,3 %). To vedlo k signifikantně vyšší expozici volným paklitaxelem u přípravku Abraxane ve srovnání s paklitaxelem založeným na bázi rozpouštědla, přestože celková expozice je srovnatelná. To je pravděpodobně způsobeno tím, že paklitaxel není zachycován Cremophor EL micelami, jak je tomu u paklitaxelu založeném na bázi rozpouštědla. Z publikované literatury vyplývá, že studie in vitro, které zkoumaly vazbu na bílkoviny lidského séra (za použití paklitaxelu v rozmezí koncentrací od 0,1 do 50 ^g/ml), naznačují, že přítomnost cimetidinu, ranitidinu, dexametazonu či difenhydraminu neovlivnila vazbu paklitaxelu na bílkoviny.
Z populační farmakokinetické analýzy vyplývá, že celkový distribuční objem je přibližně 1 741 l; velký distribuční objem naznačuje rozsáhlou extravaskulární distribuci a/nebo vazbu paklitaxelu ve tkáních.
Biotransformace a eliminace
Z publikované literatury dále vyplývá, že studie in vitro prováděné s mikrozomy lidských jater a vzorky tkání prokázaly, že paklitaxel je metabolizován především na 6a-hydroxypaklitaxel a na dva vedlejší metabolity, 3’-p-hydroxypaklitaxel a 6a-3’-p-dihydroxypaklitaxel. Tvorbu těchto hydroxylovaných metabolitů katalyzují CYP2C8, CYP3A4, respektive oba typy izoenzymů CYP2C8 i CYP3A4.
U pacientů s metastazujícím karcinomem prsu byla po 30minutové infuzi přípravku Abraxane v dávce 260 mg/m2 průměrná hodnota kumulativní exkrece nezměněné léčivé látky močí 4 % z celkové podané dávky; méně než 1 % bylo vyloučeno močí ve formě metabolitů 6a-hydroxypaklitaxelu a 3’-p-hydroxypaklitaxelu, což naznačuje rozsáhlou non renální clearance. Paklitaxel se eliminuje zejména metabolismem v játrech a biliární exkrecí.
Při klinickém rozmezí dávek 80 až 300 mg/m2 se průměrná clearance paklitaxelu v plazmě pohybuje v rozmezí 13 až 30 l/h/m2 a průměrný terminální poločas se pohybuje v rozmezí 13 až 27 hodin.
Porucha funkce jater
Vliv poruchy funkce jater na populační farmakokinetiku přípravku Abraxane byl studován u pacientů s pokročilými solidními tumory. Do této analýzy byli zahrnuti pacienti s normální funkcí jater (n = 130) a již existující mírnou (n = 8), středně těžkou (n = 7) nebo těžkou (n = 5) poruchou funkce jater (podle kritérií Organ Dysfunction Working Group NCI). Výsledky prokazují, že mírná porucha funkce jater (celkový bilirubin > 1 až < 1,5 x ULN) nemá klinicky významný vliv na farmakokinetiku paklitaxelu. U pacientů se středně těžkou (celkový bilirubin > 1,5 až < 3 x ULN) nebo těžkou (celkový bilirubin > 3 až < 5 x ULN) poruchou funkce jater dochází k poklesu maximální míry eliminace paklitaxelu o 22 % až 26 % a ke zvýšení průměrné AUC paklitaxelu přibližně o 20 % ve srovnání s pacienty s normální funkcí jater. Porucha funkce jater nemá žádný účinek na průměrnou Cmax paklitaxelu. Kromě toho je eliminace paklitaxelu nepřímo úměrná množství celkového bilirubinu a přímo úměrná množství sérového albuminu.
Farmakokinetické/farmakodynamické modelování naznačuje, že neexistuje žádná korelace mezi funkcí jater (stanovenou počáteční hladinou albuminu nebo hladinou celkového bilirubinu) a neutropenií po úpravě na expozici přípravku Abraxane.
Nejsou dostupné farmakokinetické údaje pro pacienty s celkovým bilirubinem > 5 x ULN ani pro pacienty s metastazujícím adenokarcinomem pankreatu (viz bod 4.2).
Porucha funkce ledvin
Do populační farmakokinetické analýzy byli zahrnuti pacienti s normální funkcí ledvin (n = 65) a dříve existující mírnou (n = 61), středně těžkou (n = 23) nebo těžkou (n = 1) poruchou funkce ledvin (podle kritérií FDA 2010 - draft FDA guidance criteria 2010). Mírná až středně těžká porucha funkce ledvin (clearance kreatininu > 30 až < 90 ml/min) nemá žádný klinicky významný účinek na maximální míru eliminace a systémovou expozici (AUC a Cmax) paklitaxelu. Farmakokinetické údaje pro pacienty s těžkou poruchou funkce ledvin jsou nedostatečné a pro pacienty s terminálním selháním ledvin nejsou dostupné.
Starší lidé
Do populační farmakokinetické analýzy pro přípravek Abraxane byli zahrnuti pacienti ve věkovém rozmezí od 24 do 85 let. Bylo prokázáno, že věk nemá významný vliv na maximální míru eliminace a systémovou expozici (AUC a Cmax) paklitaxelu.
Farmakokinetické/farmakodynamické modely s využitím údajů od 125 pacientů s pokročilými solidními nádory naznačuj í, že pacienti > 65 let věku mohou být náchylnější k rozvoji neutropenie během prvního léčebného cyklu, ačkoliv plazmatická expozice paklitaxelu není ovlivněna věkem.
Ostatní vnitřní faktory
Populační farmakokinetické analýzy pro přípravek Abraxane naznačují, že pohlaví, rasa (asiaté versus běloši) a typ solidního tumoru nemají klinicky významný vliv na systémovou expozici (AUC a Cmax) paklitaxelu. Pacienti s tělesnou hmotností 50 kg měli AUC paklitaxelu přibližně o 25 % nižší než ti, kteří vážili 75 kg. Klinický význam těchto zjištění je nejistý.
5.3 Předklinické údaje vztahující se k bezpečnosti
Kancerogenní potenciál paklitaxelu dosud nebyl prozkoumán. Z publikované literatury však vyplývá, že paklitaxel podávaný v klinických dávkách je potenciálně kancerogenní a genotoxický na základě farmakodynamického mechanismu jeho působení. Paklitaxel vykazuje klastogenní účinky in vitro (chromozomové aberace lidských lymfocytů) a in vivo (mikronukleární test u myší). Bylo prokázáno, že paklitaxel je genotoxický in vivo (mikronukleární test u myší), nezpůsobil však mutagenitu v Amesově testu nebo v testu genové mutace hypoxanthin-guanin fosforibosyltransferázy v buněčné linii CHO z vaječníků křečíka čínského (CHO/HGPRT).
Paklitaxel v dávkách nižších, než je léčebná dávka pro člověka, byl spojen s nízkou fertilitou a fetální toxicitou u potkanů. Studie s přípravkem Abraxane prováděné na zvířatech prokázaly nezvratné toxické účinky na samčí reprodukční orgány při klinicky relevantních hladinách expozice.
FARMACEUTICKÉ ÚDAJE
6.
6.1 Seznam pomocných látek
Roztok lidského albuminu (obsahuje sodík, natrium-oktanoát a racemický acetyltryptofan).
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřené injekční lahvičky 3 roky
Stabilita rekonstituované suspenze v injekční lahvičce
Po první rekonstituci musí být suspenze neprodleně opatrně vstříknuta do infuzního vaku. Chemická a fyzikální stabilita byla prokázána po dobu 8 hodin při teplotě 2 až 8°C v původním balení v krabičce, chráněna před ostrým světlem. Ve sterilních prostorách je možno použít alternativní ochranu před světlem.
Stabilita rekonstituované suspenze v infuzním vaku
Po rekonstituci musí být rekonstituovaná suspenze v infuzním vaku okamžitě spotřebována.
Chemická a fyzikální stabilita byla prokázána po dobu 8 hodin při teplotě do 25°C.
6.4 Zvláštní opatření pro uchovávání
Nepoužité injekční lahvičky
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem. Mráz ani chlad nemají nepříznivý vliv na stabilitu přípravku. Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Rekonstituovaná suspenze
Podmínky uchovávání tohoto léčivého přípravku po jeho rekonstituci jsou uvedeny v bodě 6.3.
6.5 Druh obalu a obsah balení
50 ml injekční lahvička (čiré sklo typu 1) se zátkou (butylová pryž) a jisticím uzávěrem (hliník) obsahuje paclitaxelum 100 mg ve formě nanočástic vázaných na albumin.
100 ml injekční lahvička (čiré sklo typu 1) se zátkou (butylová pryž) a jisticím uzávěrem (hliník) obsahuje paclitaxelum 250 mg ve formě nanočástic vázaných na albumin.
Balení obsahuje jednu injekční lahvičku.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Upozornění pro přípravu a podávání
Paklitaxel je cytotoxická protinádorová léčivá látka. Stejně jako u ostatních potenciálně toxických sloučenin je při zacházení s přípravkem Abraxane nutná zvýšená opatrnost. Doporučuje se používat rukavice, ochranné brýle a ochranný oděv. Dojde-li ke styku suspenze s pokožkou, je třeba zasažené místo neprodleně a důkladně omýt vodou a mýdlem. Jestliže dojde ke styku suspenze se sliznicemi, je nutné je důkladně opláchnout vodou. Abraxane musí připravovat a podávat pouze personál patřičně
Z důvodu možné extravazace se doporučuje v průběhu podávání přípravku pozorně sledovat místo infuze, zda nedochází k infiltraci tkání při podávání léčivého přípravku. Omezení trvání doby infuze přípravku Abraxane na 30 minut podle doporučení snižuje pravděpodobnost vzniku nežádoucích účinků spojených s infuzí.
Rekonstituce a podávání přípravku
Abraxane je dodáván jako sterilní lyofilizovaný prášek pro rekonstituci před použitím. Jeden mililitr suspenze po rekonstituci obsahuje 5 mg paklitaxelu ve formě nanočástic vázaných na albumin.
Injekční lahvička 100 mg: Sterilní injekční stříkačkou je třeba pomalu, nejméně po dobu 1 minuty, vstřikovat do injekční lahvičky s přípravkem Abraxane 20 ml infuzního roztoku chloridu sodného 9 mg/ml (0,9 %).
Injekční lahvička 250 mg: Sterilní injekční stříkačkou je třeba pomalu, nejméně po dobu 1 minuty, vstřikovat do injekční lahvičky s přípravkem Abraxane 50 ml infuzního roztoku chloridu sodného 9 mg/ml (0,9 %).
Roztok by měl dopadat na vnitřní stěnu injekční lahvičky. Roztok se nesmí vstřikovat přímo na prášek, způsobilo by to napěnění.
Po dokončení vstřikování roztoku nechte lahvičku stát nejméně 5 minut, aby se prášek důkladně navlhčil. Poté lahvičkou jemně a pomalu otáčejte a/nebo převracejte po dobu nejméně 2 minut, dokud se všechen prášek zcela nerozpustí. Dbejte, aby se nevytvořila pěna. Jestliže dojde k napěnění či shluknutí, musí se roztok nechat alespoň 15 minut odstát, než pěna opadne.
Rekonstituovaná suspenze by měla být mléčná a homogenní, bez viditelných sraženin. Může dojít k drobnému usazování rekonstituované suspenze. Jestliže jsou vidět sraženiny či usazeniny, lahvičku znovu jemně převracejte, aby se před použitím zajistila kompletní resuspenze.
Zkontrolujte, zda suspenze v injekční lahvičce neobsahuje částice. Nepodávejte rekonstituovanou suspenzi, pokud jsou v injekční lahvičce viditelné částice.
Přesný celkový objem dávky suspenze o koncentraci 5 mg/ml pro pacienta je třeba vypočítat a příslušné množství rekonstituovaného přípravku Abraxane vstříknout do prázdného sterilního vaku pro nitrožilní infuzi z PVC nebo jiného materiálu.
Použití zdravotnických prostředků obsahujících silikonový olej jako lubrikant (tj. stříkaček nebo vaků pro nitrožilní infuzi) k rekonstituci a podání přípravku Abraxane může mít za následek tvorbu bílkovinných vláken. Podávejte přípravek Abraxane pomocí infuzního setu s vestavěným 15 ^m filtrem, abyste se vyvarovali podání těchto vláken. Použití 15 ^m filtru odstraní vlákna a nezmění fyzikální ani chemické vlastnosti rekonstituovaného přípravku.
Použití filtrů s velikostí pórů menší než 15 ^m může vést k ucpání filtru.
Pro přípravu či podávání infuzí přípravku Abraxane není nutné použití speciálních nádob nebo souprav bez dioktyl-ftalátu (bis(2-ethylhexyl) ftalátu (DEHP)).
Po podání se doporučuje propláchnout nitrožilní linku injekčním roztokem chloridu sodného o koncentraci 9 mg/ml (0,9 %), aby bylo zajištěno podání celé dávky.
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Celgene Europe Limited 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/07/428/001
EU/1/07/428/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. ledna 2008
Datum posledního prodloužení registrace: 11. ledna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Celgene Europe Limited 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Držitel rozhodnutí o registraci předkládá pravidelně aktualizované zprávy o bezpečnosti pro tento léčivý přípravek v souladu s požadavky uvedenými v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a zveřejněném na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
Pokud se shodují data předložení aktualizované zprávy o bezpečnosti (PSUR) a aktualizovaného RMP, je možné je předložit současně.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
Krabička
Abraxane 5 mg/ml, prášek pro přípravu infuzní suspenze paclitaxelum
Jedna injekční lahvička obsahuje paclitaxelum 100 mg ve formě nanočástic vázaných na albumin.
Jeden ml suspenze po rekonstituci obsahuje paclitaxelum 5 mg ve formě nanočástic vázaných na albumin.
Pomocné látky: Roztok lidského albuminu (obsahuje sodík, natrium-oktanoát a racemický acetyltryptofan)
Obsahuje sodík, další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro přípravu infúzní suspenze 1 injekční lahvička 100 mg/20 ml
Před použitím si přečtěte příbalovou informaci. Intravenózní podání
Uchovávejte mimo dohled a dosah dětí.
Přípravek Abraxane nemá nahrazovat jiné formy paklitaxelu, ani jimi nemá být nahrazován.
Použitelné do:
Neotevřené injekční lahvičky: Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Po první rekonstituci: 8 hodin v chladničce v injekční lahvičce, pokud je uchovávána v krabičce, aby byl přípravek chráněn před světlem.
V infuzním vaku: až 8 hodin při teplotě do 25oC.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Celgene Europe Limited 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
EU/1/07/428/001
Č. š.:
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Injekční lahvička
Abraxane 5 mg/ml, prášek pro přípravu infuzní suspenze paclitaxelum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK_
Jedna injekční lahvička obsahuje paclitaxelum 100 mg ve formě nanočástic vázaných na albumin. Jeden ml suspenze po rekonstituci obsahuje paclitaxelum 5 mg.
3. SEZNAM POMOCNÝCH LÁTEK_
Pomocné látky: Roztok lidského albuminu (obsahuje sodík, natrium-oktanoát a racemický acetyltryptofan)
Obsahuje sodík, další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ_
Prášek pro přípravu infuzní suspenze.
1 injekční lahvička 100 mg/20 ml
Před použitím si přečtěte příbalovou informaci. Intravenózní podání
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Neotevřené injekční lahvičky: Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Celgene Europe Ltd 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
EU/1/07/428/001
Č. š.:
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Krabička
Abraxane 5 mg/ml, prášek pro přípravu infuzní suspenze paclitaxelum
Jedna injekční lahvička obsahuje paclitaxelum 250 mg ve formě nanočástic vázaných na albumin.
Jeden ml suspenze po rekonstituci obsahuje paclitaxelum 5 mg ve formě nanočástic vázaných na albumin.
Pomocné látky: Roztok lidského albuminu (obsahuje sodík, natrium-oktanoát a racemický acetyltryptofan)
Obsahuje sodík, další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Prášek pro přípravu infúzní suspenze 1 injekční lahvička 250 mg/50 ml
Před použitím si přečtěte příbalovou informaci. Intravenózní podání
Uchovávejte mimo dohled a dosah dětí.
Přípravek Abraxane nemá nahrazovat jiné formy paklitaxelu, ani jimi nemá být nahrazován.
Použitelné do:
Neotevřené injekční lahvičky: Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Po první rekonstituci: 8 hodin v chladničce v injekční lahvičce, pokud je uchovávána v krabičce, aby byl přípravek chráněn před světlem.
V infuzním vaku: až 8 hodin při teplotě do 25oC.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Celgene Europe Limited 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
EU/1/07/428/002
Č. š.:
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
Injekční lahvička
Abraxane 5 mg/ml, prášek pro přípravu infuzní suspenze paclitaxelum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK_
Jedna injekční lahvička obsahuje paclitaxelum 250 mg ve formě nanočástic vázaných na albumin. Jeden ml suspenze po rekonstituci obsahuje paclitaxelum 5 mg.
3. SEZNAM POMOCNÝCH LÁTEK_
Pomocné látky: Roztok lidského albuminu (obsahuje sodík, natrium-oktanoát a racemický acetyltryptofan)
Obsahuje sodík, další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ_
Prášek pro přípravu infuzní suspenze.
1 injekční lahvička 250 mg/50 ml
Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Použitelné do:
Neotevřené injekční lahvičky: Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Celgene Europe Ltd 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
EU/1/07/428/002
Č. š.:
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele Abraxane 5 mg/ml, prášek pro přípravu infuzní suspenze
paclitaxelum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je Abraxane a k čemu se používá
2. Čemu musíte věnovat pozornost, než Vám bude Abraxane podán
3. Jak se Abraxane používá
4. Možné nežádoucí účinky
5. Jak Abraxane uchovávat
6. Obsah balení a další informace
1. Co je Abraxane a k čemu se používá
Co je Abraxane
Léčivou látkou v přípravku Abraxane je paklitaxel navázaný na lidskou bílkovinu albumin ve formě
malých částic známých jako nanočástice. Paklitaxel patří do skupiny léčiv zvaných „taxany“, které se
používají k léčbě rakoviny.
• Paklitaxel představuje část léku působící na nádor, zastavuje dělení rakovinných buněk - buňky tedy umírají.
• Albumin je část léku, která pomáhá paklitaxelu rozpustit se v krvi a procházet přes stěnu cév do nádoru. To znamená, že není potřeba jiných chemických látek, které mohou způsobovat nežádoucí účinky, jež mohou ohrožovat život. Tyto nežádoucí účinky se vyskytují u přípravku Abraxane v podstatně menší míře.
K čemu se Abraxane používá
Abraxane se používá k léčbě těchto typů rakoviny:
Rakovina prsu
• Rakovina prsu, která se rozšířila do ostatních částí těla (nazývá se „metastatický“ karcinom prsu).
• Abraxane se používá u metastatického karcinomu prsu v případě, kdy byla vyzkoušena nejméně jedna další léčba, která ale nebyla účinná, a není pro Vás vhodná léčba obsahující skupinu léků zvaných „antracykliny“.
• Lidé s metastatickým karcinomem prsu, kterým byl podán Abraxane poté, když u nich jiná léčba selhala, častěji zaznamenávali zmenšení velikosti tumoru a žili déle než lidé, kteří podstoupili alternativní léčbu.
Rakovina slinivky břišní
• Jestliže máte metastatický karcinom slinivky břišní, používá se Abraxane společně s lékem, který se jmenuje gemcitabin. Lidé s metastatickým karcinomem slinivky břišní (nádor slinivky břišní, který se rozšířil do dalších částí těla), kteří byli v klinickém hodnocení léčeni přípravkem Abraxane s gemcitabinem, žili déle než lidé, kteří byli léčeni pouze gemcitabinem.
Rakovina plic
• Abraxane se používá také společně s lékem, který se nazývá karboplatina, jestliže máte nejčastější typ rakoviny plic, která se označuje jako „nemalobuněčný karcinom plic“.
• Abraxane se používá u nemalobuněčného karcinomu plic, když pro léčbu tohoto onemocnění není vhodná operace ani léčba ozařováním.
2. Čemu musíte věnovat pozornost, než Vám bude Abraxane podán
Nepoužívejte Abraxane,
• jestliže jste alergický/á (přecitlivělý/á) na paklitaxel nebo na kteroukoli další složku přípravku Abraxane (uvedenou v bodě 6);
• jestliže kojíte;
• jestliže máte nízký počet bílých krvinek (počet neutrofilů před léčbou <1 500 buněk/mm3 - váš lékař vás o tom bude informovat).
Upozornění a opatření
Před použitím přípravku Abraxane se poraďte se svým lékařem nebo zdravotní sestrou,
• jestliže máte špatnou funkci ledvin;
• jestliže máte závažné jaterní potíže;
• jestliže máte potíže se srdcem.
Poraďte se se svým lékařem nebo zdravotní sestrou, jestliže máte při léčbě s Abraxane jakékoli z následujících potíží. Váš lékař může ukončit léčbu nebo snížit dávkování:
• jestliže se u Vás objeví nezvyklá tvorba podlitin, krvácení nebo známky infekce, např. bolest v krku nebo horečka;
• jestliže pociťujete znecitlivění, brnění či píchání, citlivost na dotek nebo svalové slabosti;
• jestliže máte problémy s dýcháním, jako je dechová nedostatečnost nebo suchý kašel.
Děti a dospívající
Tento lék nebyl u dětí a dospívajících studován, protože se karcinom prsu, karcinom slinivky břišní a karcinom plic u těchto věkových skupin nevyskytuj í.
Další léčivé přípravky a Abraxane
Informujte svého lékaře o všech lécích, které užíváte, nebo které jste v nedávné době užíval(a), a to i o lécích, které jsou dostupné bez lékařského předpisu, včetně rostlinných přípravků. To proto, že by Abraxane mohl ovlivňovat působení některých léčivých přípravků. Některé jiné léčivé přípravky mohou také ovlivňovat působení přípravku Abraxane.
Postupujte s opatrností a poraďte se se svým lékařem, pokud užíváte Abraxane současně s kterýmikoli z následujících léčivých přípravků.
• léky k léčbě infekcí (tj. antibiotika, jako je erytromycin, rifampicin atd.; zeptejte se svého lékaře, zdravotní sestry nebo lékárníka, pokud si nejste jistý(á), zda je lék, který užíváte, antibiotikem), včetně léků k léčbě plísňových infekcí (např. ketokonazol)
• léky pomáhající stabilizovat Vaši náladu, někdy také zvané antidepresiva (např. fluoxetin)
• léky k léčbě záchvatů (epilepsie) (např. karbamazepin, fenytoin)
• léky pomáhající snižovat hladiny tuků v krvi (např. gemfibrozil)
• léky k léčbě pálení žáhy nebo žaludečních vředů (např. cimetidin)
• léky k léčbě HIV a AIDS (např. ritonavir, sachinavir, indinavir, nelfinavir, efavirenz, nevirapin) Těhotenství, kojení a plodnost
Paklitaxel může způsobit závažné vrozené vady, a proto nesmí být používán v těhotenství.
Ženy v plodném věku by měly používat účinnou antikoncepci během léčby přípravkem Abraxane a až l měsíc po jejím ukončení.
Mužům se doporučuje, aby během léčby a až šest měsíců po ní nepočali dítě a aby se před léčbou poradili ohledně konzervace spermatu, protože léčba přípravkem Abraxane může způsobit trvalou neplodnost.
Poraďte se se svým lékařem dříve, než začnete užívat tento lék.
Řízení dopravních prostředků a obsluha strojů
Někteří lidé mohou po podání přípravku Abraxane pociťovat únavu nebo závratě. Pokud tyto pocity zaznamenáte, neřiďte dopravní prostředky ani nepoužívejte stroje nebo přístroje.
Jestliže jako součást léčby dostáváte další léky, poraďte se se svým lékařem o možnosti řízení dopravních prostředků a obsluhy strojů.
Abraxane obsahuje sodík
Jeden ml Abraxane obsahuje přibližně 4,2 mg sodíku. Tuto skutečnost je třeba brát v úvahu, pokud držíte dietu s nízkým obsahem sodíku.
3. Jak se Abraxane používá
Abraxane vám bude v nitrožilní infúzi podávat lékař nebo zdravotní sestra. Dávka, kterou budete dostávat, závisí na velikosti tělesného povrchu a výsledcích krevních testů. Obvyklá dávka přípravku u karcinomu prsu je 260 mg/m2 povrchu těla podávaných po dobu 30 minut. Obvyklá dávka přípravku u pokročilého karcinomu slinivky břišní je 125 mg/m2 povrchu těla podávaných po dobu 30 minut. Obvyklá dávka přípravku u nemalobuněčného karcinomu plic je 100 mg/m2 povrchu těla podávaných po dobu 30 minut.
Jak často budete Abraxane dostávat?
Při léčbě metastatického karcinomu prsu se Abraxane obvykle podává jednou za každé tři týdny (1. den 21denního cyklu).
Při léčbě pokročilého karcinomu slinivky břišní se Abraxane podává 1., 8. a 15. den každého 28denního léčebného cyklu. Gemcitabin se podává ihned po podání Abraxane.
Při léčbě nemalobuněčného karcinomu plic se Abraxane podává jednou za každý týden (tj. 1., 8. a 15. den 21denního cyklu) a karboplatina se podává j ednou za každé tři týdny (tj. pouze 1. den každého 21denního cyklu) ihned po podání dávky Abraxane.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo zdravotní sestry.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Velmi časté nežádoucí účinky se mohou vyskytnout u více než 1 z 10 lidí:
• Vypadávání vlasů (většina případů vypadávání vlasů nastala do jednoho měsíce po zahájení léčby přípravkem Abraxane. Pokud k vypadávání vlasů dojde, je u většiny pacientů výrazné (více než 50 %))
• Vyrážka
• Abnormální snížení počtu určitých typů bílých krvinek (neutrofilů, lymfocytů nebo bílých krvinek) v krvi
• Nedostatek červených krvinek
• Snížení počtu krevních destiček v krvi
• Účinek na periferní nervy (bolest, znecitlivění, brnění nebo ztráta citu)
• Bolest v kloubu či kloubech
• Bolesti svalů
• Nevolnost, průjem, zácpa, bolestivost úst, ztráta chuti k jídlu
• Zvracení
• Dehydratace, poruchy chuti, úbytek tělesné hmotnosti
• Nízká hladina draslíku v krvi
• Deprese, potíže se spaním
• Bolest hlavy
• Zimnice
• Dýchací potíže
• Závratě
• Otoky sliznic a měkkých tkání
• Zvýšené hodnoty jaterních testů
• Bolesti končetin
• Kašel
• Bolest břicha
• Krvácení z nosu
Časté nežádoucí účinky se mohou vyskytnout až u 1 z 10 lidí:
• Svědění, suchá kůže, vady nehtů
• Infekce, horečka se sníženým počtem určitého typu bílých krvinek (neutrofilů) v krvi, návaly, kvasinková infekce, závažná infekce Vaší krve, která může být zapříčiněná sníženým počtem bílých krvinek
• Snížení počtu všech krvinek
• Bolest na hrudi nebo bolest v krku
• Trávicí potíže, nepříjemné pocity v břiše
• Ucpaný nos
• Bolest zad, bolesti kostí
• Zhoršená svalová koordinace nebo potíže při čtení, zvýšená nebo snížená produkce slz, ztráta řas
• Změny srdeční frekvence nebo rytmu, selhání srdce
• Snížený nebo zvýšený krevní tlak
• Zarudnutí nebo otok v místě vpichu jehly
• Stavy úzkosti
• Infekce v plicích
• Infekce v močových cestách
• Ucpání střeva, zánět tlustého střeva, zánět žlučovodu
• Akutní selhání ledvin
• Zvýšená hladina bilirubinu v krvi
• Vykašlávání krve
• Sucho v ústech, potíže s polykáním
• Svalová slabost
• Rozmazané vidění
Méně časté nežádoucí účinky se mohou vyskytnout až u 1 ze 100 lidí:
• Nárůst tělesné hmotnosti, zvýšená hodnota laktát dehydrogenázy v krvi, snížená funkce ledvin, zvýšená hladina cukru v krvi, zvýšená hladina fosforu v krvi
• Zhoršení nebo ztráta reflexů, mimovolní pohyby, bolest v okolí nervů, mdloby, závratě při přechodu do vzpřímené polohy, třes, ochrnutí lícního nervu
• Podráždění očí, bolest očí, zarudnutí očí, svědění očí, dvojité vidění, zhoršení vidění, mžitky, rozostřené vidění v důsledku otoku sítnice (cystoidní edém makuly)
• Bolest uší, ušní šelest
• Vykašlávání hlenu, zadýchávání se při chůzi nebo chůzi do schodů, rýma, suchý nos, oslabené dýchání, voda na plicích, ztráta hlasu, krevní sraženiny v plicích, sucho v krku
• Plynatost, žaludeční křeče, bolestivé dásně, vředy na dásních, krvácení z konečníku
• Bolestivé močení, časté močení, krev v moči, neschopnost udržet moč
• Bolest nehtů, lámavost nehtů, vypadávání nehtů, kopřivka, bolest kůže, červené spáleniny od slunce, změna barvy kůže, zvýšení pocení, pocení v noci, bílé skvrny na kůži, vředy, otoky obličeje
• Snížená hladina fosforu v krvi, zadržování tekutin, nízká hladina albuminu v krvi, zvýšená žízeň, snížená hladina vápníku v krvi, snížená hladina cukru v krvi, snížená hladina sodíku v krvi
• Bolest nebo otok v nose, kožní infekce, infekce v důsledku zavedení katétru
• Podlitiny
• Bolest v místě nádoru, odumření nádoru
• Snížení krevního tlaku při přechodu do vzpřímené polohy, studené ruce a nohy
• Obtíže při chůzi, otoky
• Alergická reakce
• Snížené funkce j ater, zvětšení j ater
• Bolest prsou
• Neklid
• Drobné krvácení do kůže v důsledku krevních sraženin
• Stav zahrnující rozpad červených krvinek a akutní selhání ledvin
Vzácné nežádoucí účinky se mohou vyskytnout až u 1 z 1 000 lidí:
• Reakce kůže na jinou látku nebo zánět plic po ozáření
• Krevní sraženiny
• Velmi pomalý pulz, srdeční infarkt
• Prosakování přípravku z žíly do tkání
• Porucha srdečního převodního systému (atrioventrikulární blokáda)
Velmi vzácné nežádoucí účinky se mohou vyskytnout až u 1 z 10 000 lidí:
• Závažný zánět/závažná vyrážka kůže a sliznic (Stevens-Johnsonův syndrom, toxická epidermální nekrolýza)
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak Abraxane uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce a na injekční lahvičce za označením Použitelné do. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Nepoužité injekční lahvičky: Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Po první rekonstituci musí být suspenze okamžitě použita. Pokud není použita okamžitě, může být suspenze uložena v chladničce (při 2 -8°C) po dobu až 8 hodin v injekční lahvičce, pokud je uchovávána v krabičce, aby byl přípravek chráněn před světlem.
Rekonstituovanou suspenzi v nitrožilní infuzi lze uchovávat až 8 hodin při teplotě do 25°C.
Za správnou likvidaci veškerého nepoužitého přípravku Abraxane zodpovídá Váš lékař nebo lékárník.
6. Obsah balení a další informace
Co Abraxane obsahuje
Léčivou látkou je paclitaxelum.
Jedna injekční lahvička obsahuje paclitaxelum 100 mg nebo 250 mg ve formě nanočástic vázaných na albumin.
Jeden ml suspenze po rekonstituci obsahuje paclitaxelum 5 mg ve formě nanočástic vázaných na albumin.
Pomocnou látkou je lidský albumin (obsahuje sodík, natrium-oktanoát a racemický acetyltryptofan). Jak Abraxane vypadá a co obsahuje toto balení
Abraxane se dodává jako bílý až žlutý prášek pro přípravu infuzní suspenze ve skleněných injekčních lahvičkách. Lahvička Abraxane obsahuje paclitaxelum 100 mg nebo 250 mg ve formě nanočástic vázaných na albumin.
Jedno balení obsahuje 1 injekční lahvičku.
Držitel rozhodnutí o registraci a výrobce
Celgene Europe Limited 1 Longwalk Road Stockley Park Uxbridge UB11 1DB Velká Británie
Další informace o tomto přípravku získáte u držitele rozhodnutí o registraci.
Tato příbalová informace byla naposledy revidována
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Zdravotničtí pracovníci
Následující informace je určena pouze pro zdravotnické pracovníky:
Pokyny pro použití, zacházení a likvidaci Upozornění pro přípravu a podávání
Paklitaxel je cytotoxická protinádorová léčivá látka. Stejně jako u ostatních potenciálně toxických sloučenin je při zacházení s přípravkem Abraxane nutná zvýšená opatrnost. Je třeba používat rukavice, ochranné brýle a oděv. Dojde-li ke styku suspenze přípravku Abraxane s pokožkou, je třeba zasažené místo neprodleně a důkladně omýt vodou a mýdlem. Jestliže dojde ke styku suspenze se sliznicemi, je nutné je důkladně opláchnout vodou. Abraxane musí připravovat a podávat pouze personál patřičně vyškolený pro zacházení s cytostatiky. S přípravkem Abraxane nesmějí pracovat těhotné ženy.
Z důvodu možné extravazace se doporučuje v průběhu podávání léčivého přípravku pozorně sledovat místo infuze, zda nedochází k infiltraci tkání při podávání přípravku. Omezení trvání doby infuze přípravku Abraxane na 30 minut podle doporučení snižuje pravděpodobnost vzniku nežádoucích účinků spojených s infuzí.
Abraxane je dodáván jako sterilní lyofilizovaný prášek pro rekonstituci před použitím. Jeden mililitr suspenze po rekonstituci obsahuje 5 mg paklitaxelu ve formě nanočástic vázaných na albumin. Rekonstituovaná suspenze přípravku Abraxane se podává nitrožilně pomocí infuzního setu s vestavěným 15 pm filtrem.
Rekonstituce 100 mg:
Sterilní injekční stříkačkou je třeba nejméně po dobu 1 minuty pomalu vstřikovat do 100 mg injekční lahvičky s přípravkem Abraxane 20 ml infuzního roztoku chloridu sodného 9 mg/ml (0,9 %).
Rekonstituce 250 mg:
Sterilní injekční stříkačkou je třeba nejméně po dobu 1 minuty pomalu vstřikovat do 250 mg injekční lahvičky s přípravkem Abraxane 50 ml infuzního roztoku chloridu sodného 9 mg/ml (0,9 %).
Roztok by měl dopadat na vnitřní stěnu injekční lahvičky. Roztok se nesmí vstřikovat přímo na prášek, způsobilo by to napěnění.
Po dokončení vstřikování roztoku nechte lahvičku stát nejméně 5 minut, aby se prášek důkladně navlhčil. Poté lahvičkou jemně a pomalu otáčejte a/nebo převracejte po dobu nejméně 2 minut, dokud se všechen prášek zcela nerozpustí. Dbejte, aby se nevytvořila pěna. Jestliže dojde k napěnění či shluknutí, je třeba nechat suspenzi alespoň 15 minut odstát, než pěna opadne.
Rekonstituovaná suspenze by měla být mléčná a homogenní, bez viditelných sraženin. Může dojít k drobnému usazování rekonstituované suspenze. Jestliže jsou vidět sraženiny či usazeniny, lahvičku znovu jemně převracejte, aby se před použitím zajistila kompletní resuspenze.
Zkontrolujte, zda suspenze v injekční lahvičce neobsahuje částice. Nepodávejte rekonstituovanou suspenzi, pokud jsou v injekční lahvičce viditelné částice.
Přesný celkový objem dávky suspenze o koncentraci 5 mg/ml pro pacienta je třeba vypočítat a příslušné množství rekonstituovaného přípravku Abraxane vstříknout do prázdného sterilního vaku pro nitrožilní infuzi z PVC nebo jiného materiálu.
Použití zdravotnických prostředků obsahujících silikonový olej jako lubrikant (tj. stříkaček nebo vaků pro nitrožilní infuzi) k rekonstituci a podání přípravku Abraxane může mít za následek tvorbu bílkovinných vláken. Podávejte přípravek Abraxane pomocí infuzního setu s vestavěným 15 pm filtrem, abyste se vyvarovali podání těchto vláken. Použití 15 pm filtru odstraní vlákna a nezmění fyzikální ani chemické vlastnosti rekonstituovaného přípravku.
Použití filtrů s velikostí pórů menší než 15 pm může vést k ucpání filtru.
Pro přípravu či podávání infuzí přípravku Abraxane není nutné použití speciálních nádob nebo souprav bez ftalátů DEHP.
Po podání se doporučuje propláchnout nitrožilní linku injekčním roztokem chloridu sodného o koncentraci 9 mg/ml (0,9 %), aby bylo zajištěno podání celé dávky.
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. Stabilita
Neotevřené injekční lahvičky přípravku Abraxane jsou stabilní do data vyznačeného na obalu, jestliže jsou uchovávány v krabičce, aby byl přípravek chráněn před světlem. Mráz ani chlad nemají nepříznivý vliv na stabilitu přípravku. Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Stabilita rekonstituované suspenze v injekční lahvičce
Po první rekonstituci musí být suspenze neprodleně opatrně vstříknuta do infuzního vaku. Chemická a fyzikální stabilita byla prokázána po dobu 8 hodin při teplotě 2 až 8°C v původní krabičce chráněna před ostrým světlem.
Stabilita rekonstituované suspenze v infuzním sáčku
Po rekonstituci musí být rekonstituovaná suspenze v infúzním vaku okamžitě spotřebována. Byla prokázána chemická a fyzikální stabilita po dobu 8 hodin při teplotě do 25°C.
48