Emend 125 Mg + 80 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
EMEND 40 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tobolka obsahuje aprepitantum 40 mg.
Pomocná látka se známým účinkem Jedna tobolka obsahuje 40 mg sacharózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tobolky jsou matné s bílým tělem a hořčicově žlutým víčkem; na těle je cirkulárně černým inkoustem vytištěno „464“ a „40 mg“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
EMEND 40 mg je indikován k prevenci pooperační nevolnosti a zvracení (postoperative nauzea and vomiting, PONV) u dospělých.
4.2 Dávkování a způsob podání
Dávkování
Pokud se týče potřeby profylaktické léčby pooperační nevolnosti a zvracení (PONV), je nutno vzít v úvahu doporučení pro léčbu v klinické praxi.
Doporučená perorální dávka přípravku EMEND je jedna 40mg dávka podaná během 3 hodin před indukcí anestézie.
Zvláštní populace Starší osoby (> 65 let)
U starších osob není nutno dávku nijak upravovat (viz bod 5.2).
Pohlaví
S ohledem na pohlaví není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin ani u pacientů v terminálním stádiu renálního onemocnění podstupujících hemodialýzu není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou poruchou funkce jater není nutno dávku nijak upravovat. Pokud se týče pacientů se středně těžkou poruchou funkce jater, je k dispozici pouze omezené množství dat, přičemž u pacientů s těžkou poruchou funkce jater nejsou k dispozici žádné údaje. Těmto pacientům se aprepitant musí podávat opatrně (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku EMEND 40 mg u dětí a dospívajících mladších 18 let nebyla dosud stanovena. V současnosti dostupné údaje jsou popsány v bodech 5.1 a 5.2, nicméně žádná doporučení ohledně dávkování nemohou být dána.
Způsob podání
Tvrdou tobolku je nutno polknout vcelku.
Přípravek EMEND lze užívat bez ohledu na jídlo.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Pacienti se středně těžkou až těžkou poruchou funkce jater
Pro pacienty se středně těžkou poruchou funkce jater existuje pouze omezené množství dat a žádné údaje nejsou k dispozici pro pacienty s těžkou poruchou funkce jater. U těchto pacientů je nutno přípravek EMEND používat s maximální opatrností (viz bod 5.2).
Interakce na CYP3A4
EMEND (40 mg) je nutno podávat s opatrností pacientům současně užívajícím pimozid, terfenadin, astemizol, cisaprid nebo deriváty námelových alkaloidů. Inhibice izoenzymu 3A4 cytochromu P450 (CYP3A4) aprepitantem může vyvolat zvýšení plazmatických koncentrací těchto léčivých látek, případně způsobit závažné nežádoucí reakce (viz bod 4.5).
Současné podávání s hormonální antikoncepcí
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a ještě 2 měsíce po poslední dávce přípravku EMEND je třeba používat alternativní nehormonální záložní antikoncepční metody (viz bod 4.5).
Ohledně dalších informací o možných interakcích aprepitantu ve vyšších a násobných dávkách viz prosím souhrn údajů o přípravku pro EMEND 80 mg tvrdé tobolky a pro EMEND 125 mg tvrdé tobolky.
Pomocné látky
Přípravek EMEND tobolky obsahuje sacharózu. Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy, malabsorpcí glukózy-galaktózy nebo deficitem sacharáza-izomaltázy nemají tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aprepitant je substrát a na dávce závislý inhibitor a induktor CYP3A4. Aprepitant je rovněž induktorem CYP2C9. Během léčby působí jedna 40mg dávka aprepitantu doporučená pro PONV slabou inhibici CYP3A4. Po léčbě způsobuje přípravek EMEND dočasnou mírnou indukci CYP2C9, CYP3A4 a glukuronidace. Aprepitant byl zkoušen při vyšších dávkách. Během léčby nevolnosti a zvracení vyvolaných chemoterapií (CINV) představoval 3denní režim s dávkami 125 mg/80 mg aprepitantu středně silný inhibitor CYP3A4. Nezdá se, že by aprepitant interagoval s P-glykoproteinovým transportérem, jak naznačuje nepřítomnost interakce aprepitantu s digoxinem.
Účinek aprepitantu na farmakokinetiku dalších léčivých látek Inhibice CYP3A4
Jako slabý inhibitor CYP3A4 může aprepitant (40 mg) zvýšit plazmatické koncentrace současně perorálně podávaných léčivých látek, které se metabolizují cestou CYP3A4. Celková expozice perorálně podávaným substrátům CYP3A4 se může zvýšit přibližně 1,5krát po jedné 40 mg dávce aprepitantu; předpokládá se, že účinek aprepitantu na plazmatické koncentrace intravenózně podávaných substrátů CYP3A4 bude menší.
EMEND 40 mg je nutno podávat s opatrností pacientům užívajícím pimozid, terfenadin, astemizol, cisaprid nebo deriváty námelových alkaloidů. Inhibice CYP3A4 aprepitantem může vyvolat zvýšení plazmatických koncentrací těchto léčivých látek, případně způsobit závažné reakce.
Kortikosteroidy
Dexamethason: Jedna 40mg dávka aprepitantu při podání spolu s jednou perorální 20 mg dávkou dexamethasonu zvýšila hodnotu AUC dexamethasonu 1,45 krát. Nedoporučuje se dávku nijak upravovat.
Methylprednisolon: Přestože současné podávání methylprednisolonu s jednou 40mg dávkou aprepitantu nebylo hodnoceno, vyvolává jedna 40mg dávka aprepitantu slabou inhibici CYP3A4 a nepředpokládá se, že by změnila plazmatické koncentrace methylprednisolonu v klinicky významné míře. Proto se úprava dávky nedoporučuje.
Midazolam
Hodnota AUC perorálně podávaného midazolamu se po podání jedné 40 mg dávky aprepitantu spolu s jednou perorální 2mg dávkou midazolamu zvýšila 1,2 krát; tento účinek nebyl hodnocen jako klinicky významný.
Indukce
Jako mírný induktor CYP2C9, CYP3A4 a glukuronidace může v průběhu dvou týdnů po zahájení léčby aprepitant snižovat plazmatické koncentrace substrátů eliminovaných těmito cestami.
U substrátů CYP2C9 a CYP3A4 je indukce přechodná, přičemž maximálního účinku se dosáhne po 3 až 5 dnech. Tento účinek může být zachován po dobu několika dní, přičemž se předpokládá, že za dva týdny po ukončení léčby přípravkem EMEND bude klinicky nevýznamný. Údaje týkající se účinků CYP2C8 a CYP2C19 chybí. Současné podávání přípravku EMEND s léčivými látkami, o nichž je známo, že se metabolizují prostřednictvím CYP2C9 (např. fenytoin, warfarin), může vést k nižším plazmatickým koncentracím těchto léčivých látek. Na základě interakčních studií s tolbutamidem a perorální antikoncepcí může být celková expozice současně podávaným léčivým látkám metabolizovaným prostřednictvím CYP2C9 nebo CYP3A4 snížena až o 15 až 30 %.
Hormonální antikoncepce
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a 2 měsíce po poslední dávce přípravku je nutno používat alternativní nehormonální záložní antikoncepční metody.
Antagonisté 5-HT3
V klinických studiích lékových interakcí nevykazoval aprepitant žádné klinicky významné účinky na farmakokinetiku ondansetronu, granisetronu nebo hydrodolasetronu (aktivní metabolit dolasetronu).
Účinky jiných léčivých přípravků na farmakokinetiku aprepitantu
K současnému podávání přípravku EMEND s léčivými látkami, které inhibují aktivitu CYP3A4 (např. ketokonazolem, itrakonazolem, vorikonazolem, posakonazolem, klarithromycinem, telithromycinem, nefazodonem a inhibitory proteázy), je třeba přistupovat opatrně, protože se předpokládá, že tato kombinace povede k několikanásobným zvýšením plazmatických koncentrací aprepitantu (viz bod 4.4).
Je nutno se vyvarovat současného podávání přípravku EMEND s léčivými látkami, které silně indukují aktivitu CYP3A4 (např. s rifampicinem, fenytoinem, karbamazepinem, fenobarbitalem), protože tato kombinace vede ke snížení plazmatických koncentrací aprepitantu, což může vést ke snížení účinnosti přípravku EMEND. Současné podávání přípravku EMEND s bylinnými přípravky obsahujícími třezalku tečkovanou (Hypericumperforatum) se nedoporučuje.
Ketokonazol
Jestliže se podal aprepitant v jediné 125mg dávce 5. den 10denního léčebného režimu se silným inhibitorem CYP3A4 ketokonazolem podaným v dávce 400 mg/den, zvýšila se hodnota AUC aprepitantu přibližně 5krát a střední hodnota terminálního poločasu aprepitantu se zvýšila přibližně 3krát.
Rifampicin
Jestliže se podal aprepitant v jediné 375 mg dávce 9. den 14denního léčebného režimu se silným induktorem CYP3A4 rifampicinem podaným v dávce 600 mg/den, snížila se hodnota AUC aprepitantu o 91 % a střední hodnota terminálního poločasu aprepitantu se snížila o 68 %.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a 2 měsíce po poslední dávce přípravku EMEND je nutno používat alternativní nehormonální záložní antikoncepční metody (viz body 4.4 a 4.5).
Nejsou dostupné žádné klinické údaje týkající se těhotenství vystavených aprepitantu. Ve studiích se zvířaty se neobjevily žádné známky přímých nebo nepřímých škodlivých účinků na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Možné účinky na reprodukci změn v regulaci neurokininů nejsou známy. Přípravek EMEND by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Aprepitant se vylučuje do mléka kojících potkanů. Není známo, zda se aprepitant vylučuje do lidského mateřského mléka; proto se kojení během léčby přípravkem EMEND nedoporučuje.
Fertilita
Studie fertility neukazují žádné přímé nebo nepřímé škodlivé účinky pokud jde o páření, fertilitu, embryonální/fetální vývoj či počty spermií a jejich motilitu (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek EMEND může mít mírný vliv na schopnost řídit nebo obsluhovat stroje. Po užití přípravku EMEND se mohou objevit závratě a únava (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Profil bezpečnosti aprepitantu byl hodnocen přibližně u 6 500 dospělých.
Nežádoucí účinky byly hlášeny přibližně u 4 % dospělých léčených aprepitantem v dávce 40 mg ve srovnání s přibližně 6 % pacientů, kteří dostávali ondansetron intravenózně v dávce 4 mg.
V kontrolovaných klinických studiích u dospělých, jimž byla podána celková anestézie, dostalo 564 pacientů perorální aprepitant v dávce 40 mg a 538 pacientů dostalo ondansetron intravenózně v dávce 4 mg. Většina nežádoucích účinků popisovaných v těchto klinických studiích byly mírné až střední intenzity.
Nejčastější nežádoucí reakcí s vyšším výskytem u dospělých léčených aprepitantem v dávce 40 mg (1,1 %) než u pacientů s ondansetronem (1,0 %) bylo zvýšení hodnot ALT.
Tabulkový seznam nežádoucích účinků
Následující nežádoucí účinky byly pozorovány ve studiích PONV u dospělých léčených režimem s aprepitantem s vyšší frekvencí než při terapii ondansetronem nebo po uvedení přípravku na trh:
Frekvence jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000) a velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Frekvence |
|
Poruchy imunitního systému |
hypersenzitivní reakce včetně anafylaktických reakcí |
není známo |
|
Psychiatrické poruchy |
insomnie |
méně časté |
|
Poruchy nervového systému |
dysartrie, hypestézie, poruchy čití |
méně časté |
|
Poruchy oka |
mióza, snížení ostrosti vidění |
méně časté |
|
Srdeční poruchy |
bradykardie |
méně časté |
|
Respirační, hrudní a mediastinální poruchy |
dušnost, sípot |
méně časté |
|
Gastrointestinální poruchy |
bolest horní poloviny břicha, abnormální střevní zvuky, sucho v ústech, nauzea, žaludeční dyskomfort, zácpa*, subileus* |
méně časté |
|
Poruchy kůže a podkožní tkáně |
svědění, vyrážka, kopřivka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza |
není známo |
|
Vyšetření |
zvýšení ALT |
časté |
* Hlášeno u pacientů užívajících vyšší dávky aprepitantu.
Popis vybraných nežádoucích účinků
U dospělých léčených aprepitantem v režimu 125 mg/80 mg kvůli nevolnosti a zvracení vyvolaných chemoterapií (CINV), byly pozorovány následující další nežádoucí účinky s výskytem častějším než při standardní léčbě: abdominální extenze, bolest břicha, akné, anémie, úzkost, zvýšení AST, asténie, zvýšení alkalické fosfatázy v krvi, snížení sodíku v krvi, kandidóza, kardiovaskulární porucha, nepříjemné pocity na hrudi, poruchy vnímání, konjunktivitida, kašel, snížení chuti k jídlu, dezorientace, závrať, perforace duodenálního vředu, poruchy vnímání chutí, dyspepsie, dysurie, říhání, eufórická nálada, tvrdá stolice, únava, febrilní neutropenie, flatulence, poruchy chůze, gastroezofageální refluxní nemoc, přítomnost glukózy v moči, škytavka, návaly horka, hyperhidróza,letargie, celkový pocit nemoci, svalové spasmy, svalová slabost, nauzea*, neutropenická kolitida, pokles počtu neutrofilů, otoky, bolest v orofaryngu, palpitace, fotosenzitivní reakce, polakisurie, polydipsie, postnazální zatékání hlenu, svědivá vyrážka, pozitivní test na červené krvinky v moči, seborea, kožní léze, kýchání, ospalost, stafylokoková infekce, stomatitida, podráždění v krku, tinnitus, zvýšená tvorba moči, zvracení*, úbytek hmotnosti.
*Nausea a zvracení byly parametry účinnosti v prvních 5 dnech následujících po chemoterapeutické léčbě a byly hlášeny jako nežádoucí účinky pouze poté.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování je nutno EMEND vysadit a zajistit obecnou podpůrnou léčbu a sledování pacienta. Vzhledem k antiemetickému účinku aprepitantu může snaha o vyvolání zvracení pomocí léčivých přípravků selhat.
Aprepitant nelze odstranit hemodialýzou.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiemetika a antinauzeancia, ATC kód: A04AD12
Aprepitant je selektivní antagonista s vysokou afinitou pro receptory neurokininu 1 (NKj) lidské substance P.
Ve dvou multicentrických, randomizovaných, dvojitě zaslepených klinických studiích fáze III u dospělých s účinnou srovnávací látkou a s paralelními skupinami byl aprepitant srovnáván s ondansetronem z hlediska PONV u 1 658 pacientů podstupujících otevřenou operaci břišní dutiny. Většina dospělých byly ženy (> 90 %), převážně po prodělané gynekologické operaci. Pacienti byli randomizováni do skupin, kdy dostávali 40 mg aprepitantu, 125 mg aprepitantu nebo 4 mg ondansetronu. Aprepitant byl podáván perorálně s 50 ml vody 1 až 3 hodiny před anestézií. Ondansetron se aplikoval intravenózně těsně před indukcí anestézie. Antiemetický účinek aprepitantu byl hodnocen v období 0 až 48 hodin po skončení operace.
Výsledky prokazují, že vyšší procento dospělých po operaci vykáže kompletní odpověď na léčbu (bez zvracení a bez použití „záchranné“ terapie) s aprepitantem 40 mg než s ondatrosetronem 4 mg (spodní mez C.I. 0,0 udává hraniční významnost), jak je popsáno v Tabulce 1
Tabulka 1
Procento dospělých po operaci s odpovědí na léčbu podle léčebných skupin Kombinované výsledky ze 2 studií fáze III
|
Aprepitan 40 mg per (N = 54 |
tum orálně U) |
Ondansetron 4 mg intravenózně (N = 526) |
Procentuální rozdíl (%)§ a 95% C.I.# | |||
|
n/m |
(%) |
n/m |
(%) |
% |
95% C.I. | |
|
Kompletní odpověď (0-24 hodině |
298/541 |
(55.1) |
258/526 |
(49.0) |
5.9 |
(0.0, 11.8) |
^ Kompletní odpověď: bez zvracení a bez použití „záchranné" terapie § Rozdíl (%) vypočítaný jako aprepitant 40 mg minus Ondansetron 4 mg
# Rozdíl (%) a 95% interval spolehlivosti vypočítaný s použitím stratifikované metody dle Miettinen-
Nurminena s použitím vážení dle Cochran-Mantel-Haenszela
Snížení rizika případu zvracení za období 0 až 24 hodin s aprepitantem 40 mg vzhledem k ondansetronu 4 mg bylo 53,3 % (95% interval spolehlivosti: 35,3 až 66,3) v analýze, která vyhodnocovala pacienty v okamžiku použití „záchranné" terapie.
Pediatrická populace
Bezpečnost a exploratorní účinnost aprepitantu byly hodnoceny v klinické studii fáze I (N = 50) využívající 40 mg prášku pro perorální suspenzi. Procento subjektů, které během prvních 24 hodin po zákroku nehlásily žádné zvracení, bylo u subjektů léčených aprepitantem podobné jako u subjektů léčených ondansetronem. Nebyla identifikována žádná nová bezpečnostní rizika. Údaje z této malé studie však nepodporují závěry ohledně optimálního dávkovacího režimu. Další studie hodnotící podávání aprepitantu pediatrickým pacientům probíhají (ohledně pediatrického podávání viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Farmakokinetika aprepitantu má nelineární charakter. Clearance i absolutní biologická dostupnost se se zvyšováním dávky snižují.
Absorpce
Průměrná absolutní biologická dostupnost perorálně podaného aprepitantu činí 67 % u 80mg tobolky a 59 % u 125mg tobolky. Průměrné maximální plazmatické koncentrace (Cmax) aprepitantu byly dosaženy přibližně po 4 hodinách (tnax).
Po perorálním podání jedné 40mg dávky přípravku EMEND nalačno dosáhla hodnota AUC0-<X)
(průměr ± SD) 8,0 ±2,1 ^g • h/ml a hodnota Cmax dosáhla 0,7 ± 0,24 ^g/ml. Medián tmax byl
3,0 hodiny.
Současné užívání 40mg dávky při standardní snídani snížilo pouze hodnotu Cmax aprepitantu o 18 % bez ovlivnění hodnoty AUC. Toto nebylo považováno za klinicky významné.
Distribuce
Aprepitant se ve vysoké míře váže na proteiny, se střední hodnotou 97 %. Geometrická střední hodnota zdánlivého distribučního objemu v rovnovážném stavu (Vdss) je u člověka přibližně 66 litrů.
Biotransformace
Aprepitant prochází rozsáhlou biotransformací. U zdravých mladých dospělých jedinců vykazuje aprepitant po dobu 72 hodin po jednorázové intravenózní aplikaci 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, v plazmě přibližně 19 % radioaktivity, což ukazuje na značnou přítomnost metabolitů v plazmě. V lidské plazmě bylo zjištěno dvanáct metabolitů aprepitantu. Metabolismus aprepitantu probíhá ve velké míře cestou oxidace v morfolinovém kruhu a jeho postranních řetězcích a výsledné metabolity jsou pouze slabě aktivní. In vitro studie s lidskými jaterními mikrozómy ukázaly, že aprepitant se biotransformuje primárně cestou CYP3A4, případně s malým podílem CYP1A2 a CYP2C19.
Eliminace
Aprepitant se nevylučuje močí v nezměněné podobě. Metabolity se vylučují močí a žlučí ve stolici.
Po jednorázově intravenózně aplikované dávce 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, zdravým jedincům bylo 57 % radioaktivity zjištěno v moči a 45 % ve stolici.
Plazmatická clearance aprepitantu závisí na dávce, se zvyšující se dávkou se snižuje a v rozmezí terapeutických dávek se pohybovala přibližně na hodnotách od 60 do 72 ml/min. Terminální poločas byl přibližně 9 hodin po podání jedné 40mg dávky.
Farmakokinetika u speciálních populací
Starší osoby: Po perorálním podání jedné 125mg dávky aprepitantu 1. den a dávky 80 mg jednou denně 2. až 5. den byla hodnota AUC aprepitantu 1. den o 21 % vyšší a 5. den o 36 % vyšší u starších jedinců (> 65 let) ve srovnání s mladšími dospělými. Hodnota Cmax byla u starších ve srovnání s mladšími dospělými 1. den o 10 % vyšší a 5. den o 24 % vyšší. Tyto rozdíly se nepovažovaly za klinicky významné. U starších pacientů není třeba dávku nijak upravovat.
Pohlaví: Po perorálním podání jediné 125mg dávky aprepitantu byla hodnota Cmax aprepitantu u žen ve srovnání s muži o 16 % vyšší. Poločas aprepitantu je u žen ve srovnání s muži o 25 % nižší a k dosažení tmax dochází zhruba ve stejnou dobu. Tyto rozdíly nejsou považovány za klinicky významné. Dávku přípravku EMEND není nutno podle pohlaví pacienta nijak upravovat.
Porucha funkce jater: Mírná porucha funkce jater (Childovo-Pughovo hodnocení, třída A) farmakokinetiku aprepitantu v klinicky významné míře neovlivňuje. U pacientů s mírnou poruchou funkce jater není třeba dávkování nijak upravovat. Z dostupných dat nelze činit žádné závěry ohledně vlivu středně těžké poruchy funkce jater (Childovo-Pughovo hodnocení, třída B) na farmakokinetiku aprepitantu. K dispozici nejsou žádné klinické ani farmakokinetické údaje od pacientů s těžkou poruchou funkce jater (Childovo-Pughovo hodnocení, třída C).
Porucha funkce ledvin: Pacientům s těžkou poruchou funkce ledvin (CrCl < 30 ml/min) a pacientům s terminálním renálním onemocněním (end stage renal disease, ESRD) s potřebou hemodialýzy byla podána jednorázová 240mg dávka aprepitantu.
U pacientů s těžkou poruchou funkce ledvin se hodnota AUC0-<X) celkového aprepitantu (nevázaného i vázaného na proteiny) ve srovnání se zdravými jedinci snížila o 21 % a hodnota Cmax se snížila o 32 %. U pacientů s ESRD podstupujících hemodialýzu se hodnota AUC0-<X) celkového aprepitantu snížila o 42 % a hodnota Cmax se snížila o 32 %. Vzhledem k mírnému poklesu vazby aprepitantu na proteiny u pacientů s renálním onemocněním nebyla hodnota AUC farmakologicky aktivní části nenavázaného léku u pacientů s poruchou funkce ledvin ve srovnání se zdravými jedinci významně ovlivněna. Hemodialýza prováděná 4 nebo 48 hodin po podání dávky neměla na farmakokinetiku aprepitantu významný účinek; v dialyzátu bylo zjištěno méně než 0,2 % dávky.
U pacientů s poruchou funkce ledvin ani u pacientů s ESRD podstupujících hemodialýzu není zapotřebí dávkování přípravku EMEND nijak upravovat.
Pediatrická populace: Ve studii využívající prášek pro perorální suspenzi vedla jednorázová dávka 40 mg aprepitantu podávaná dospívajícím pacientům (ve věku 12 až 17 let) ke střední hodnotě AUC0-48hod 6 pg/ml se střední hodnotou maximální plazmatické koncentrace (Cmax) 0,5 pg/ml, kterých se dosáhlo za přibližně 4 hodiny. Podávání dávek upravených podle plochy povrchu těla pacientům ve věku 6 měsíců až méně než 12 let vedlo ke střední hodnotě AUC0-48hod nad 4 pg/ml se střední hodnotou Cmax nad 0,5 pg/ml, jichž se dosáhlo za přibližně 3 hodiny.
Vztah mezi koncentrací a účinkem
Studie využívající pozitronovou emisní tomografii (PET) s použitím vysoce specifického značení receptorů NK u zdravých mladých mužů ukázaly, že aprepitant prostupuje do mozku a obsazuje receptory NK v závislosti na dávce a koncentraci v plazmě. Předpokládá se, že plazmatické koncentrace aprepitantu dosažené při 3-denním léčebném režimu s přípravkem EMEND zajišťují více než 95% obsazení receptorů NK v mozku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické údaje získané na základě konvenčních studií toxicity, genotoxicity, hodnocení kancerogenního potenciálu, reprodukční a vývojové toxicity po jednorázovém a opakovaném podání přípravku neodhalily žádné zvláštní riziko pro člověka. Je nutno mít na paměti, že systémová expozice u dospělých samců potkanů byla nižší, než byla terapeutická expozice u člověka při dávce 40 mg.
Z toho plyne, že žádné dostačující vyhodnocení týkající se možného účinku na plodnost samců potkanů nebylo provedeno. Nicméně v 9-měsíční studii u psů nedošlo ke změně hmotnosti žádného orgánu ani k žádným histomorfologickým nálezům, které by se objevily v reprodukčních orgánech samců vystavených 35-násobné dávce nad terapeutickou expozici 40 mg u lidí. Přestože v reprodukčních studiích, pokud byla dospělá zvířata samičího pohlaví vystavena expozici 3,5 až 4krát vyšší než byly terapeutické expozice u člověka při dávce 40 mg, nebyly zaznamenány žádné nežádoucí účinky, nejsou známy možné vlivy na reprodukci při změnách v regulaci neurokininu.
Ve studii juvenilní toxicity na potkanech, kterým se podával aprepitant od 10. do 63. postnatálního dne, docházelo u samic k časnému vaginálnímu otevírání od dávek 250 mg/kg dvakrát denně a u samců k opožděné prepuciální separaci, a to od dávek 10 mg/kg dvakrát denně. Nebyl zde žádný odstup od klinicky relevantní expozice. Nedošlo k žádným účinkům souvisejícím s léčbou ohledně páření, fertility či embryonálního/fetálního vývoje a nevyskytly se žádné patologické změny reprodukčních orgánů. Ve studii juvenilní toxicity na psech ošetřovaných od 14. do 42. postnatálního dne byla u samců při dávce 6 mg/kg/den pozorována snížená hmotnost varlat a velikost Leydigových buněk a u samic při dávkách od 4 mg/kg/den byla pozorována zvýšená hmotnost dělohy, hypertrofie dělohy a krčku a otok vaginálních tkání. Nebyl zde žádný odstup od klinicky relevantní expozice aprepitantu. Při krátkodobé léčbě podle doporučeného dávkovacího režimu se klinická relevance těchto zjištění nepovažuje za pravděpodobnou.
6.1 Seznam pomocných látek
Obsah tobolky Sacharóza
Mikrokrystalická celulóza (E 460)
Hyproloza (E 463)
Natrium-lauryl-sulfát
Obal tobolky Želatina
Oxid titaničitý (E 171)
Žlutý oxid železitý (E 172)
Inkoust na potisk Šelak
Hydroxid draselný Černý oxid železitý (E 172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
K dispozici jsou různé velikosti balení.
Hliníkový blistr obsahující jednu 40mg tobolku.
5 hliníkových blistrů, každý obsahující jednu 40mg tobolku.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO
EU/1/03/262/008
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. listopadu 2003 Datum prodloužení registrace: 22. září 2008
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
EMEND 165 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tobolka obsahuje aprepitantum 165 mg.
Pomocná látka se známým účinkem
Jedna tvrdá tobolka obsahuje 165 mg sacharózy.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
Tobolky jsou neprůhledné se světle modrým víčkem a bílým tělem s „466“ a „165 mg“ vytištěným černým inkoustem radiálně na jedné straně těla a logem Merck na druhé straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence akutní a pozdní nevolnosti a zvracení u dospělých v souvislosti s vysoce emetogenní protinádorovou chemoterapií založenou na bázi cisplatiny.
Prevence nevolnosti a zvracení u dospělých v souvislosti se středně emetogenní protinádorovou chemoterapií.
EMEND 165 mg se podává jako součást kombinované terapie (viz bod 4.2).
4.2 Dávkování a způsob podání
Dávkování
Přípravek EMEND 165 mg se podává pouze 1. den, přibližně jednu hodinu před chemoterapií jako součást režimu zahrnujícího kortikosteroid a antagonistu 5-HT3.
K prevenci nevolnosti a zvracení v souvislosti s emetogenní protinádorovou chemoterapií se u dospělých doporučují následující režimy:
Vysoce emetogenní chemoterapeutický režim
|
1. den |
2. den |
3. den |
4. den | |
|
EMEND |
165 mg perorálně |
žádný |
žádný |
žádný |
|
Dexamethason |
12 mg perorálně |
8 mg perorálně |
8 mg perorálně dvakrát denně |
8 mg perorálně dvakrát denně |
|
Antagonisté 5-HT3 |
Standardní dávka antagonistů 5- ht3. Ohledně příslušného dávkování |
žádný |
žádný |
žádný |
|
zvoleného antagonisty 5-HT3 viz informace o daném přípravku |
Dexamethason se musí podávat 1. den 30 minut před zahájením chemoterapie a 2. až 4. den ráno. Dexamethason se rovněž musí podávat 3. a 4. den večer. Dávka dexamethasonu přispívá k interakcím léčivých látek.
Středně emetogenní chemoterapeutický režim
|
1. den | |
|
EMEND |
165 mg perorálně |
|
Dexamethason |
12 mg perorálně |
|
Antagonisté 5-HT3 |
Standardní dávka antagonistů 5-HT3. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku |
Dexamethason se musí podávat 1. den 30 minut před zahájením chemoterapie. Dávka dexamethasonu přispívá k interakcím léčivých látek.
Množství údajů o účinnosti při kombinaci s jinými kortikosteroidy a antagonisty 5-HT3 je omezeno. Další informace o současném podávání s kortikosteroidy jsou uvedeny v bodu 4.5. U současně podávaných léčivých přípravků obsahujících antagonistu 5-HT3 nahlédněte do jejich souhrnu informací o přípravku.
Jako jediná dávka je rovněž k dispozici fosaprepitant 150 mg, což je lyofilizované proléčivo aprepitantu k intravenóznímu podání, přičemž tento lék lze použít jako alternativu k perorálnímu přípravku EMEND 165 mg.
Zvláštní _ populace Starší osoby (> 65 let)
U starších osob není nutno dávku nijak upravovat (viz bod 5.2).
Pohlaví
S ohledem na pohlaví není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin ani u pacientů v terminálním stádiu renálního onemocnění podstupujících hemodialýzu není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou poruchou funkce jater není nutno dávku nijak upravovat. U pacientů se středně těžkou poruchou funkce jater je k dispozici pouze omezené množství dat a nejsou dostupné žádné údaje od pacientů s těžkou poruchou funkce jater. Těmto pacientům se aprepitant musí podávat opatrně (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku EMEND 165 mg u dětí a dospívajících mladších 18 let nebyla stanovena. K dispozici nejsou žádné údaje. U této populace mohou být vhodnější jiné lékové formy/síly.
Způsob podání
Tvrdou tobolku je nutno polknout vcelku.
Přípravek EMEND lze užívat bez ohledu na jídlo.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Současné podávání s pimozidem, terfenadinem, astemizolem nebo cisapridem (viz bod 4.5).
4.4 Zvláštní upozornění a opatření pro použití Pacienti se středně těžkou až těžkou poruchou funkce jater
Pro pacienty se středně těžkou poruchou funkce jater existuje pouze omezené množství dat a žádné údaje nejsou k dispozici pro pacienty s těžkou poruchou funkce jater. U těchto pacientů je nutno přípravek EMEND používat s opatrností (viz bod 5.2).
Interakce na CYP3A4
EMEND je nutno podávat s opatrností pacientům současně užívajícím léčivé látky, které se primárně metabolizují cestou CYP3A4 a které mají úzké terapeutické rozmezí, jako je cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, deriváty námelových alkaloidů fentanyl a chinidin (viz bod 4.5). Navíc je nutno k podávání současně s irinotekanem přistupovat s obzvláštní opatrností, protože uvedená kombinace by mohla mít za následek zvýšenou toxicitu.
Současné podávání s warfarinem (substrát CYP2C9)
U pacientů dlouhodobě léčených warfarinem je nutno hodnotu mezinárodního normalizovaného poměru (INR) pozorně sledovat během léčby přípravkem EMEND a po dobu 14 dnů po podání přípravku EMEND (viz bod 4.5).
Současné podávání s hormonální antikoncepcí
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a 2 měsíce po poslední dávce přípravku EMEND je třeba používat alternativní nehormonální záložní antikoncepční metody (viz bod 4.5).
Pomocné látky
Tobolky přípravku EMEND obsahují sacharózu. Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy, malabsorpcí glukózy-galaktózy nebo deficitem sacharáza-izomaltázy nemají tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aprepitant je substrát, středně silný inhibitor a induktor CYP3A4. Aprepitant je také induktor CYP2C9. Během léčby přípravkem EMEND je CYP3A4 inhibován po dobu až 4 dní. Přípravek EMEND způsobuje přechodnou mírnou indukci CYP2C9, CYP3A4 a glukuronidace přetrvávající asi jeden týden po ukončení léčby. Nezdá se, že by aprepitant interagoval s P-glykoproteinovým transportérem, jak naznačuje nepřítomnost interakce aprepitantu s digoxinem.
Účinek aprepitantu na farmakokinetiku dalších léčivých látek Inhibice CYP3A4
Jako středně silný inhibitor CYP3A4 může aprepitant zvýšit plazmatické koncentrace současně podávaných léčivých látek, které se metabolizují cestou CYP3A4. Celková expozice perorálně podávaným substrátům CYP3A4 se může po jediné 165mg dávce přípravku EMEND zvýšit na dobu 2 dní přibližně až 3krát, přičemž po dávce přípravku EMEND 165 mg klesne na výchozí hodnoty za asi 4 dny. Předpokládá se, že účinek aprepitantu na plazmatické koncentrace intravenózně podávaných substrátů CYP3A4 bude menší. Přípravek EMEND se nesmí podávat současně s pimozidem, terfenadinem, astemizolem nebo cisapridem (viz bod 4.3). Inhibice CYP3A4 aprepitantem by mohla mít za následek zvýšené plazmatické koncentrace uvedených léčivých látek, což může vyvolat závažné nebo život ohrožující reakce. Při současném podávání přípravku EMEND a léčivých látek, které jsou metabolizovány převážně prostřednictvím CYP3A4 a které mají úzké terapeutické rozmezí, jako je cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, diergotamin, ergotamin, fentanyl a chinidin, se doporučuje opatrnost (viz bod 4.4).
Kortikosteroidy
Dexamethason: interakční studie s aprepitantem v dávce 165 mg a dexamethasonem nebyly provedeny; nicméně při podávání přípravku EMEND 165 mg a perorálního dexamethasonu je nutno vzít v potaz následující studii s 200 mg aprepitantu. Aprepitant, pokud se podává jako jediná dávka 200 mg v systém stavu (standardní lehká snídaně) 1. den s perorálním dexamethasonem, podávaným perorálně v dávce 12 mg 1. den a v dávce 8 mg 2. až 4. den, zvyšoval AUC0-24hod dexamethasonu 1. a 2. den 2,1- až 2,3krát, menší měrou (1,4násobný vzestup) 3. den a 4. den neměl žádný vliv (1,1násobek). Denní dávka dexamethasonu 1. a 2. den se musí při současném podávání 1. den s přípravkem EMEND 165 mg snížit přibližně o 50 %, aby se dosáhlo expozice dexamethasonu podobné expozici dosahované při podávání bez přípravku EMEND 165 mg.
Methylprednisolon: interakční studie s aprepitantem v dávce 165 mg a methylprednisolonem nebyly provedeny; nicméně při podávání přípravku EMEND 165 mg s methylprednisolonem je nutno vzít v potaz následující studii s režimem podávání aprepitantu v režimu 125 mg/80 mg. Pokud se EMEND podával v léčebném režimu v dávce 125 mg první den a 80 mg/den 2. a 3. den, zvýšil hodnotu AUC methylprednisolonu, který je substrátem CYP3A4, 1. den 1,3krát a 3. den 2,5krát v případě, že se methylprednisolon podával současně intravenózně v dávce 125 mg 1. den a perorálně v dávce 40 mg 2. a 3. den.
V kontinuální léčbě methylprednisolonem se může hodnota AUC methylprednisolonu koncem 14denního období po zahájení podávání přípravku EMEND snížit v důsledku indukčního účinku aprepitantu na CYP3A4. Lze očekávat, že tento účinek bude u perorálně podávaného methylprednisolonu výraznější.
Chemoterapeutické léčivé přípravky
Interakční studie s aprepitantem v dávce 165 mg a chemoterapeutickými léčivými přípravky nebyly provedeny; nicméně na základě studií třídenního režimu perorálního aprepitantu a docetaxelu a vinorelbinu se nepředpokládá, že by přípravek EMEND měl klinicky relevantní interakce s intravenózně podávaným docetaxelem a vinorelbinem. Ve farmakokinetických studiích neměl přípravek EMEND při podávání v režimu 125 mg 1. den a 80 mg/den 2. a 3. den vliv na farmakokinetiku docetaxelu podaného intravenózně 1. den nebo vinorelbinu podaného intravenózně 1. nebo 8. den. Protože účinek přípravku EMEND na farmakokinetiku perorálně podávaných substrátů CYP3A4 je větší než účinek přípravku EMEND na farmakokinetiku intravenózně podávaných substrátů CYP3A4, nelze vyloučit interakce s perorálně podávanými chemoterapeutickými léčivými přípravky, které se metabolizují převážně nebo částečně cestou CYP3A4 (např. etoposid, vinorelbin). U pacientů dostávajících léčivé přípravky, které se metabolizují především nebo i částečně CYP3A4, se doporučuje opatrnost, přičemž může být vhodné tyto pacienty dodatečně sledovat (viz bod 4.4). Po uvedení na trh byly hlášeny při současném podávání aprepitantu a ifosfamidu případy neurotoxicity, potenciální nežádoucí účinek ifosfamidu.
Imunosupresiva
Po jediné 165 mg dávce aprepitantu se předpokládá přechodné střední zvýšení trvající 2 dny případně následované mírným poklesem expozice imunosupresivům metabolizovaným prostřednictvím CYP3A4 (např. cyklosporin, takrolimus, everolimus a sirolimus). S ohledem na krátké trvání zvýšené expozice se v den podání přípravku EMEND 165 mg a den poté snížení dávky imunosupresiva založené na monitorování terapeutické dávky imunosupresiva nedoporučuje.
Midazolam
Interakční studie s aprepitantem v dávce 165 mg a midazolamem nebyly provedeny; nicméně při podávání přípravku EMEND 165 mg a léčivých přípravků metabolizovaných prostřednictvím CYP3A4 je nutno vzít v potaz následující studii s 200 mg aprepitantu. Ve studii se 2 mg midazolamu podávaného perorálně spolu s 200 mg aprepitantu v sytém stavu (standardní lehká snídaně) bylaAUC0-o, midazolamu, což je citlivý substrát CYP3A4, 1. den zvýšena 3,2krát. 4. den nebyly pozorovány žádné klinicky významné účinky (1,2násobné zvýšení AUC0-o midazolamu) a 8. den byla pozorována mírná změna AUC0-o midazolamu (35% pokles).
Při současném podávání midazolamu nebo jiných benzodiazepinů metabolizovaných cestou CYP3A4 (alprazolam, triazolam) spolu s přípravkem EMEND 165 mg je nutno vzít v úvahu možné účinky zvýšených plazmatických koncentrací těchto léčivých přípravků.
Indukce
Jako mírný induktor CYP2C9, CYP3A4 a glukuronidace může aprepitant snižovat plazmatické koncentrace substrátů eliminovaných těmito cestami. Tento účinek může být zřejmý přibližně 7 dní po podání jediné 165mg dávky přípravku EMEND. Tento účinek se udržuje několik dní, poté pomalu klesá a během 14 dní po ukončení léčby přípravkem EMEND je klinicky nevýznamný. Jediná dávka aprepitantu 200 mg 1. den k níž byl 1., 4. a 8. den podán midazolam, což je citlivý substrát CYP3A4, vedla 8. den k 35% snížení AUC0-o midazolamu. Předpokládá se, že přípravek EMEND 165 mg by mol způsobit podobnou indukci CYP2C9, CYP3A4 a glukuronidace, jakou způsobuje podávání třídenního perorálního režimu aprepitantu, kdy byla pozorována přechodná indukce s maximálním účinkem 6 až 8 dní po první dávce aprepitantu. Třídenní perorální režim aprepitantu vedl k přibližně 30 až 35% snížení AUC substrátů CYP2C9 a až k 64% poklesu minimálních koncentrací ethinylestradiolu. Ohledně účinků na CYP2C8 a CYP2C19 údaje chybějí. Pokud se s přípravkem EMEND 165 mg podává warfarin, acenokumarol, tolbutamid, fenytoin nebo jiné léčivé látky, o nichž je známo, že se metabolizují prostřednictvím CYP2C9, doporučuje se opatrnost.
Warfarin
U pacientů dlouhodobě léčených warfarinem je třeba pozorně sledovat protrombinový čas (INR) během léčby a po dobu 14 dnů po použití přípravku EMEND 165 mg k léčbě chemoterapií vyvolané nevolnosti a zvracení (viz bod 4.4). Jestliže se podala jediná 125mg dávka přípravku EMEND 1. den a dávka 80 mg/den 2. a 3. den zdravým jedincům ve stabilizovaném stavu při dlouhodobé léčbě warfarinem, neměl EMEND 3. den žádný účinek na hodnotu plazmatické AUC pro R(+) ani S(-) warfarin, nicméně 5 dní po skončení podávání přípravku EMEND ale došlo ke 34% snížení dolní hodnoty koncentrace S(-) warfarinu (substrátu CYP2C9) spolu se 14% poklesem hodnoty INR.
Tolbutamid
Pokud se EMEND podával v dávce 125 mg 1. den a v dávce 80 mg/den 2. a 3. den, snížil 4. den hodnotu AUC tolbutamidu (substrátu CYP2C9) o 23 %, 8. den o 28 %, a 15. den o 15 % v případě, že se jediná 500mg dávka tolbutamidu podala perorálně před zahájením 3denní kúry s přípravkem EMEND a pak 4., 8., a 15. den.
Hormonální antikoncepce
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a ještě 2 měsíce po poslední dávce přípravku EMEND je třeba používat alternativní nehormonální záložní antikoncepční metody.
V klinické studii byly 1. až 21. den podávány jednorázové dávky perorální antikoncepce obsahující ethinylestradiol a norethisteron spolu s přípravkem EMEND, podávaným v režimu v dávce 125 mg 8. den a 80 mg/den 9. a 10. den s ondansetronem v dávce 32 mg podaným intravenózně 8. den
a dexamethasonem v dávce 12 mg podaným perorálně 8. den a dále 8 mg/den 9., 10. a 11. den. V této studii se minimální koncentrace ethinylestradiolu snížily 9. až 21. den až o 64 % a minimální koncentrace norethisteronu až o 60 %.
Antagonisté 5-HT3
V klinických studiích lékových interakcí nevykazoval aprepitant, pokud se podával v režimu 125 mg 1. den a 80 mg 2. a 3. den, žádné klinicky významné účinky na farmakokinetiku ondansetronu, granisetronu nebo hydrodolasetronu (aktivní metabolit dolasetronu).
Účinky jiných léčivých přípravků na farmakokinetiku aprepitantu
K současnému podávání přípravku EMEND s léčivými látkami, které inhibují aktivitu CYP3A4 (např. ketokonazolem, itrakonazolem, vorikonazolem, posakonazolem, klarithromycinem, telithromycinem, nefazodonem a inhibitory proteázy), je třeba přistupovat opatrně, protože se předpokládá, že tato kombinace povede k několikanásobnému zvýšení plazmatické koncentrace aprepitantu (viz bod 4.4).
Je nutno se vyvarovat současného podávání přípravku EMEND s léčivými látkami, které silně indukují aktivitu CYP3A4 (např. s rifampicinem, fenytoinem, karbamazepinem, fenobarbitalem), protože tato kombinace vede ke snížení plazmatických koncentrací aprepitantu, což může vést ke snížení účinnosti přípravku EMEND. Současné podávání přípravku EMEND s bylinnými přípravky obsahujícími třezalkou tečkovanou (Hypericumperforatum) se nedoporučuje.
Ketokonazol
Jestliže se podal aprepitant v jediné 125mg dávce 5. den 10denního léčebného režimu se silným inhibitorem CYP3A4 ketokonazolem podaným v dávce 400 mg/den, zvýšila se hodnota AUC aprepitantu přibližně 5krát a střední hodnota terminálního poločasu aprepitantu se zvýšila přibližně 3krát.
Rifampicin
Jestliže se podal aprepitant v jediné 375 mg dávce 9. den 14denního léčebného režimu se silným induktorem CYP3A4 rifampicinem podaným v dávce 600 mg/den, snížila se hodnota AUC aprepitantu o 91 % a střední hodnota terminálního poločasu aprepitantu se snížila o 68 %.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a 2 měsíce po poslední dávce přípravku EMEND je nutno používat alternativní nehormonální záložní antikoncepční metody (viz body 4.4 a 4.5).
Nejsou dostupné žádné klinické údaje týkající se těhotenství vystavených aprepitantu. Potenciální reprodukční toxicita aprepitantu nebyla zcela stanovena, protože míry expozice vyšší než hodnoty terapeutické expozice u člověka při dávkách 125 mg/80 mg a 165 mg nebylo možno ve studiích se zvířaty dosáhnout. Tyto studie nenaznačily žádné přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Možné účinky změn v regulaci neurokininů na reprodukci nejsou známy. Přípravek EMEND se nesmí podávat během těhotenství, pokud to není nezbytně nutné.
Kojení
Aprepitant se vylučuje do mléka kojících potkanů. Není známo, zda se aprepitant vylučuje do lidského mateřského mléka, a proto se kojení během léčby přípravkem EMEND nedoporučuje.
Fertilita
Potenciál aprepitantu navodit účinky na fertilitu nebyl dosud plně popsán, protože expozičních hladin přesahujících terapeutickou expozici u lidí nemohlo být ve studiích na zvířatech dosaženo. Tyto studie fertility neukazují na přímé nebo nepřímé škodlivé účinky ohledně páření, fertility, embryonálního/fetálního vývoje či počtu a motility spermií (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek EMEND může mít mírný vliv na schopnost řídit nebo obsluhovat stroje. Závratě a únava se mohou objevit po podání přípravku EMEND (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnostní profil aprepitantu byl hodnocen u přibližně 6 500 dospělých.
Na základě srovnatelného farmakokinetického/farmakodynamického profilu se předpokládá, že jednodenní perorální režim přípravku EMEND 165 mg bude mít podobný bezpečnostní profil a profil snášenlivosti jako je tomu u jednodenního režimu fosaprepitantu v dávce 150 mg a u třídenního perorálního režimu aprepitantu u pacientů na chemoterapii (viz bod 5.2).
Nejčastějšími nežádoucími účinky u dospělých léčených vysoce emetogenní chemoterapií, které byly hlášeny s vyšší incidencí u pacientů léčených v třídenním režimu s perorálním aprepitantem než při standardní terapii byly: škytavka (4,6 % oproti 2,9 %), zvýšení alaninaminotransferázy (ALT) (2,8 % oproti 1,1 %), dyspepsie (2,6 % oproti 2,0 %), zácpa (2,4 % oproti 2,0 %), bolest hlavy (2,0 % oproti
1,8 %) a pokles chuti k jídlu (2,0 % oproti 0,5 %). Nejčastějším nežádoucím účinkem hlášeným s vyšší incidencí u pacientů léčených třídenním režimem s perorálním aprepitantem než při standardní terapii u pacientů léčených středně emetogenní chemoterapií byla únava (1,4 % oproti 0,9 %).
Tabulkový seznam nežádoucích účinků
Následující nežádoucí účinky byly pozorovány v souhrnné analýze ve studiích vysoce a středně emetogenní chemoterapie nebo po uvedení na trh s vyšší incidencí u aprepitantu než u standardní léčby dospělých:
Četnosti jsou definovány jako: velmi časté (> 1/10); časté (> 1/100 až < 1/10); méně časté (> 1/1 000 až < 1/100); vzácné (> 1/10 000 až < 1/1 000) a velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Frekvence |
|
Infekce a infestace |
kandidóza, stafylokokové infekce |
vzácné |
|
Poruchy krve a lymfatického systému |
febrilní neutropenie, anemie |
méně časté |
|
Poruchy imunitního systému |
hypersenzitivní reakce včetně anafylaktických reakcí |
není známo |
|
Poruchy metabolismu a výživy |
snížení chuti k jídlu |
časté |
|
polydipsie |
vzácné | |
|
Psychiatrické poruchy |
méně časté | |
|
dezorientace, euforická nálada |
vzácné | |
|
Poruchy nervového systému |
časté | |
|
závrať, somnolence |
méně časté | |
|
kognitivní poruchy, letargie, poruchy vnímání chutí |
vzácné | |
|
Poruchy oka |
konjunktivitida |
vzácné |
|
Poruchy ucha a labyrintu |
tinnitus |
vzácné |
|
Srdeční poruchy |
palpitace |
méně časté |
|
bradykardie, kardiovaskulární porucha |
vzácné | |
|
Cévní poruchy |
méně časté | |
|
Respirační, hrudní a mediastinální poruchy |
škytavka |
časté |
|
bolest v orofaryngu, kýchání, kašel, postnazální zatékání hlenu (postnasal drip), podráždění hrdla |
vzácné | |
|
Gastrointestinální poruchy |
zácpa, dyspepsie |
časté |
|
říhání, nauzea*, zvracení*, gastroesofageální refluxní choroba, bolest břicha, sucho v ústech, flatulence |
méně časté |
|
Třída orgánových systémů |
Nežádoucí účinek |
Frekvence |
|
perforace duodenálního vředu, stomatitida, abdominální distenze, tvrdá stolice, neutropenická kolitida |
vzácné | |
|
Poruchy kůže a podkožní tkáně |
vyrážka, akné |
méně časté |
|
fotosenzitivita, hyperhidróza, seborea, kožní léze, svědivá vyrážka, Stevens-Johnsonův syndrom |
vzácné | |
|
svědění, kopřivka |
není známo | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
svalová slabost, svalové spasmy |
vzácné |
|
Poruchy ledvin a močových cest |
méně časté | |
|
polakisurie |
vzácné | |
|
Celkové poruchy a reakce v místě aplikace |
časté | |
|
astenie, malátnost |
méně časté | |
|
edém, nepříjemný pocit na hrudi, poruchy chůze |
vzácné | |
|
Vyšetření |
zvýšení ALT |
časté |
|
zvýšení AST, zvýšení alkalické fosfatázy |
méně časté | |
|
pozitivní test na červené krvinky v moči, zvýšení hladiny sodíku v krvi, pokles tělesné hmotnosti, pokles počtu neutrofilů, přítomnost glukózy v moči, zvýšená tvorba moči |
vzácné |
* Nauzea a zvracení byly parametry účinnosti v prvních 5 dnech léčby po chemoterapii a pouze potom byly hlášeny jako nežádoucí účinky.
Popis vybraných nežádoucích účinků
Profily nežádoucích reakcí u dospělých v prodloužených studiích s vysoce nebo středně emetogenní chemoterapií s opakovanými až 6 dalšími cykly chemoterapie byly celkově podobné jako profily pozorované v 1. cyklu.
V další klinické studii kontrolované léčivou látkou, prováděné u 1 169 dospělých pacientů kterým byl podáván třídenní režim perorálního aprepitantu a vysoce emetogenní chemoterapie, byl profil nežádoucích účinků celkově podobný jako profily pozorované v jiných studií vysoce emetogenní terapie s třídenním režimem perorálního aprepitantu.
U pacientů léčených aprepitantem kvůli pooperační nauzee a zvracení (PONV) byly pozorovány další nežádoucí účinky, které měly vyšší incidenci než u ondansetronu: bolest v horní části břicha, abnormální střevní zvuky, zácpa*, dysartrie, dušnost, hypestézie, nespavost, mióza, nauzea, poruchy čití, žaludeční dyskomfort, subileus*, snížená ostrost vidění, sípání.
* Hlášeno u pacientů užívajících vyšší dávky aprepitantu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování je nutno přípravek EMEND vysadit a poskytnout obecnou podpůrnou terapii a sledování pacienta. Vzhledem k antiemetogennímu účinku aprepitantu může snaha o vyvolání zvracení podáním léčivého přípravku selhat.
Aprepitant nelze odstranit hemodialýzou.
FARMAKOLOGICKÉ VLASTNOSTI
5.
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiemetika a léčiva k terapii nauzey, ATC kód: A04AD12.
Aprepitant je selektivní antagonista s vysokou afinitou k receptorům neurokinin 1 (NKj) humánní substance P. Fosaprepitant je proléčivo aprepitantu a po intravenózním podání je rychle konvertován na aprepitant.
Na základě srovnatelného farmakokinetického/farmakodynamického profilu se předpokládá, že jednodenní perorální režim přípravku EMEND 165 mg bude mít podobný profil účinnosti jako je tomu u jednodenního režimu fosaprepitantu v dávce 150 mg a u třídenního perorálního režimu aprepitantu (viz bod 5.2).
Třídenní režim podávání aprepitantu u dospělých
Ve dvou randomizovaných, dvojitě zaslepených studiích zahrnujících celkem 1 094 dospělých pacientů léčených chemoterapií zahrnující cisplatinu v dávce > 70 mg/m2, byl režim aprepitant v kombinaci s ondansetronem/dexamethasonem (viz bod 4.2) srovnáván se standardním režimem (placebo plus ondansetron 32 mg aplikovaný intravenózně 1. den plus dexamethason 20 mg perorálně podaný 1. den a 8 mg perorálně dvakrát denně 2. až 4. den). I když se v klinických hodnoceních používala 32mg intravenózní dávka ondansetronu, nejedná se již o doporučenou dávku. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku.
Účinnost se hodnotila pomocí následujícího souhrnného parametru: úplná odpověď (definovaná jako žádné emetické epizody, bez použití záchranné léčby) primárně v 1. cyklu. Výsledky se hodnotily v každé studii jednotlivě a pak pro tyto 2 studie dohromady.
Přehled klíčových výsledků studie z kombinované analýzy je uveden v Tabulce 1.
Tabulka 1
Procento dospělých pacientů léčených vysoce emetogenní chemoterapií s odpovědí na léčbu podle
|
léčebných skupin a fáze - 1. cyklus | |||
|
SLOŽENÁ KRITÉRIA |
Režim |
Standardní |
Rozdíly* |
|
s aprepitantem |
terapie | ||
|
(N = 521)t |
(N = 524)T | ||
|
% |
% |
% (95% CI) | |
|
Úplná odpověď (bez zvracení a záchranné léčby) | |||
|
Celkem (0-120 hodin) |
67,7 |
47,8 |
19,9 (14,0-25,8) |
|
0-24 hodin |
86,0 |
73,2 |
12,7 (7,9-17,6) |
|
25-120 hodin |
71,5 |
51,2 |
20,3 (14,5-26,1) |
JEDNOTLIVÁ KRITÉRIA
Bez zvracení (žádné emetické epizody bez ohledu na použití záchranné léčby)
|
Celkem (0-120 hodin) 0-24 hodin 25-120 hodin |
71,9 86,8 76,2 |
49,7 74,0 53,5 |
22,2 12,7 22,6 |
(16,4-28,0) (8,0-17,5) (17,0-28,2) |
|
Bez významné nevolnosti (maximálně VAS < 25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
72,1 |
64,9 |
7,2 |
(1,6-12,8) |
|
25-120 hodin |
74,0 |
66,9 |
7,1 |
(1,5-12,6) |
* Intervaly spolehlivosti byly vypočítány bez korekce na pohlaví a souběžně podávanou chemoterapii, které byly zahrnuty do primární analýzy podílu šancí (odds ratio) a logistických modelů. t Jeden pacient v režimu pouze s aprepitantem měl údaje v akutní fázi a byl vyloučen z celkové analýzy i z analýzy prodloužené fáze; jeden pacient pouze ve standardním režimu měl údaje v prodloužené fázi a byl vyloučen z celkové analýzy i z analýzy akutní fáze.
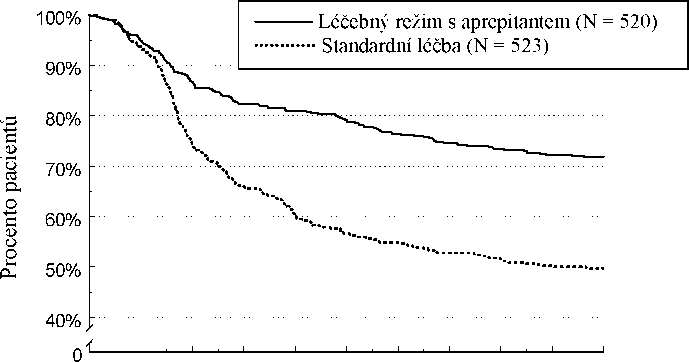
Odhad doby do první epizody zvracení v kombinované analýze je zobrazen vynesením hodnot metodou Kaplana-Meiera v Grafu 1.
Graf 1
Procento dospělých pacientů léčených vysoce emetogenní chemoterapií, u nichž nedošlo k epizodě zvracení - 1. cyklus
0 12 24 36 48 60 72 84 96 108 120
Čas (v hodinách)
Statisticky významné rozdíly v účinnosti byly pozorovány v každé z těchto 2 jednotlivých studií.
V těchto dvou klinických studiích 851 dospělých pacientů pokračovalo v prodloužené studii
s opakovanými až 5 dalšími cykly chemoterapie. Účinnost režimu s aprepitantem zůstala zachována během všech cyklů.
V randomizované, dvojitě zaslepené studii s celkovým počtem 866 dospělých pacientů (864 žen,
2 muži) léčených chemoterapií, která zahrnovala cyklofosfamid 750 až 1 500 mg/m2 nebo cyklofosfamid 500 až 1 500 mg/m2 a doxorubicin (<60 mg/m2) nebo epirubicin (<100 mg/m2), byl režim aprepitant v kombinaci s ondansetronem/dexamethasonem (viz bod 4.2) srovnáván se standardní terapií (placebo plus ondansetron 8 mg perorálně (dvakrát 1. den a každých 12 hodin 2. a 3. den) plus dexamethason 20 mg perorálně 1. den).
Účinnost se hodnotila pomocí souhrnného parametru: úplná odpověď (definovaná jako žádné emetické epizody a nepoužití záchranné léčby) primárně během 1. cyklu.
Přehled hlavních výsledků studie je uveden v Tabulce 2.
Tabulka 2
Procento dospělých pacientů odpovídajících na léčbu podle léčebné skupiny a fáze - 1. cyklus
|
SLOŽENÁ |
Režim |
Standardní |
Rozdíly* |
|
KRITÉRIA |
s aprepitantem |
terapie | |
|
(N = 433)t |
(N = 424) | ||
|
% |
% |
% (95% CI) |
|
lná odpověď (bez zvracení a záchranné terapie) | ||||
|
Celkem (0-120 hodin) |
50,8 |
42,5 |
8,3 |
(1,6-15,0) |
|
0-24 hodin |
75,7 |
69,0 |
6,7 |
(0,7-12,7) |
|
25-120 hodin |
55,4 |
49,1 |
6,3 |
(-0,4-13,0) |
|
JEDNOTLIVÁ KRITÉRIA | ||||
|
z zvracení (žádné emetické epizody bez ohledu na |
použití záchranné terapie) | |||
|
Celkem (0-120 hodin) |
75,7 |
58,7 |
17,0 |
(10,8-23,2) |
|
0-24 hodin |
87,5 |
77,3 |
10,2 |
(5,1-15,3) |
|
25-120 hodin |
80,8 |
69,1 |
11,7 |
(5,9-17,5) |
|
z významné nevolnosti (maximální |
VAS < 25 mm na stupnici 0-100 mm) | |||
|
Celkem (0-120 hodin) |
60,9 |
55,7 |
5,3 |
(-1,3-11,9) |
|
0-24 hodin |
79,5 |
78,3 |
1,3 |
(-4,2-6,8) |
|
25-120 hodin |
65,3 |
61,5 |
3,9 |
(-2,6-10,3) |
* Intervaly spolehlivosti byly vypočítány bez korekce na věkovou kategorii (< 55 let, > 55 let) a skupiny hodnotících lékařů, které byly zahrnuty do primární analýzy podílu šancí (odds ratio) a logistických modelů. t Jeden pacient pouze v režimu s aprepitantem měl údaje z akutní fáze a byl vyloučen z celkové analýzy i z analýzy prodloužené fáze.
Středně emetogenní chemoterapie
Ve stejné klinické studii pokračovalo 744 dospělých pacientů v prodloužené studii s opakovanými až 3 dalšími cykly chemoterapie. Účinnost režimu s aprepitantem zůstala zachována během všech cyklů.
Ve druhé multicentrické, randomizované, dvojitě zaslepené studii s paralelními skupinami byl režim s aprepitantem porovnáván se standardní léčbou u 848 dospělých pacientů (652 žen, 196 mužů), kterým byl podáván chemoterapeutický režim, který zahrnoval jakékoli intravenózní dávky oxaliplatiny, karboplatiny, epirubicinu, idarubicinu, ifosfamidu, irinotekanu, daunorubicinu, doxorubicinu; cyklofosfamid intravenózní (< 1 500 mg/m2) nebo cytarabin intravenózně (> 1 g/m2). Pacientům, kterým byl podáván režim s aprepitantem, byla chemoterapie podávána na různé typy nádorů, včetně 52 % s rakovinou prsu, 21 % s gastrointestinálními rakovinami včetně kolorektálního karcinomu, 13 % s rakovinou plic a 6 % s gynekologickými rakovinami. Režim s aprepitantem v kombinaci s režimem ondansetron/dexamethason (viz bod 4.2) byl porovnáván se standardní léčbou (placebo v kombinaci s ondansetronem 8 mg perorálně (dvakrát 1. den a každých 12 hodin 2. a 3. den) plus dexamethason 20 mg perorálně 1. den).
Účinnost byla založena na vyhodnocení následujících primárních a klíčových sekundárních koncových parametrů: žádné zvracení za celou dobu (0 až 120 hodin po chemoterapii), vyhodnocení bezpečnosti a snášenlivosti režimu s aprepitantem při chemoterapií vyvolané nevolnosti a zvracení (CINV) a úplná odpověď (definovaná jako žádné zvracení a žádné použití záchranné léčby) za celou dobu (0 až 120 hodin po chemoterapii). Navíc byla sledována, jako exploratorní koncový parametr, „Žádná významná nauzea“ (No Significant Nausea) za celou dobu (0 až 120 hodin po chemoterapii), jak v akutní, tak i v prodloužené fázi formou post-hoc analýzy.
Souhrn klíčových výsledků studie je uveden v Tabulce 3.
Tabulka 3
P rocento dospělých pacientů odpovídajících na léčbu podle léčebné skupiny a fáze pro studii 2 -
cyklus 1
středně emetogenní chemoterapie
|
Režim s aprepi- |
Standardní |
Rozdíly | |
|
tantem |
léčba | ||
|
(N = 425) |
3 II O Os | ||
|
% |
0/ % |
0/ % |
(95% CI) |
Úplná odpověď (žádné zvracení a žádná záchranná léčba)
|
Celkem (0-120 hodin) 0-24 hodin 25-120 hodin |
68.7 89,2 70.8 |
56.3 80.3 60,9 |
12,4 8.9 9.9 |
(5,9, 18,9) (4,0, 13,8) (3,5, 16,3) |
|
Žádná emeze (žádné emetické epizody bez ohledu na použití záchranné léčby) | ||||
|
Celkem (0-120 hodin) |
76,2 |
62,1 |
14,1 |
(7,9, 20,3) |
|
0-24 hodin |
92,0 |
83,7 |
8,3 |
(3,9, 12,7) |
|
25-120 hodin |
77,9 |
66,8 |
11,1 |
(5,1, 17,1) |
|
Žádná významná nauzea (maximální VAS <25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
73,6 |
66,4 |
7,2 |
(1,0, 13,4) |
|
0-24 hodin |
90,9 |
86,3 |
4,6 |
(0,2, 9,0) |
|
25-120 hodin |
74,9 |
69,5 |
5,4 |
(-0,7, 11,5) |
*Intervaly spolehlivosti byly vypočteny bez úpravy podle pohlaví a oblasti, které byly zařazeny v primární analýze za použití logistických modelů.
Přínos kombinované terapie s aprepitantem u celé hodnocené populace byl dán zejména výsledky pozorovanými u pacientů se slabou kontrolou při standardním režimu, jako jsou ženy, i když výsledky byly numericky lepší bez ohledu na věk, typ tumoru nebo pohlaví. Úplné odpovědi bylo dosaženo u 209/324 (65 %) v případě režimu s aprepitantem a u 161/320 (50 %) při standardní terapii u žen a u mužů šlo v případě režimu s aprepitantem o 83/101 (82 %) a při standardní terapii o 68/87 (78 %).
Jednodenní režim fosaprepitantu 150 mg u dospělých
V randomizované, dvojitě zaslepené studii s aktivní kontrolou a paralelními skupinami byl porovnáván fosaprepitant v dávce 150 mg (N = 1 147) s třídenním režimem aprepitantu (N = 1 175) u dospělých pacientů léčených režimem vysoce emetogenní chemoterapie, který zahrnoval cisplatinu (> 70 mg/m2). Režim fosaprepitantu sestával z fosaprepitantu v dávce 150 mg 1. den v kombinaci intravenózním ondansetronem v dávce 32 mg 1. den a dexamethasonem v dávce 12 mg 1. den a 8 mg 2. den a 8 mg dvakrát denně 3. a 4. den. Režim aprepitantu sestával z aprepitantu 125 mg 1. den a 80 mg/den 2. a 3. den v kombinaci s intravenózním ondansetronem v dávce 32 mg 1. den a dexamethasonem v dávce 12 mg 1. den a 8 mg denně 2. až 4. den. K zaslepení se použilo placebo fosaprepitantu, aprepitantu a dexamethasonu (večer 3. a 4. den) (viz bod 4.2). I když se v klinických hodnoceních používala 32mg intravenózní dávka ondansetronu, nejedná se již o doporučenou dávku. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku.
Účinnost byla založena na vyhodnocení následujících souhrnných parametrů: úplná odpověď jak za celou dobu, tak v pozdních fázích a žádné zvracení za celou dobu. Bylo prokázáno, že fosaprepitant v dávce 150 mg není horší než třídenní režim aprepitantu. Souhrn primárních a sekundárních kritérií hodnocení je uveden v Tabulce 4.
Tabulka 4
P rocento dospělých pacientů odpovídajících na léčbu léčených vysoce emetogenní chemoterapií
podle léčené skupiny a fáze — 1. cyklus
|
KRITÉRIA HODNOCENÍ* |
Režim s fosaprepi-tantem (N = 1 106)** % |
Režim s aprepitantem (N = 1 134)** % |
Rozdíl1 (95% CI) |
|
Úplná odpověď1 | |||
|
Celkem § |
71,9 |
72,3 |
-0,4 (-4,1, 3,3) |
|
Pozdní fáze§§ |
74,3 |
74,2 |
0,1 (-3,5, 3,7) |
|
Žádné zvracení | |||
|
Celkem§ |
72,9 |
74,6 |
-1,7 (-5,3, 2,0) |
* Primární koncový parametr je tučně.
** N: počet pacientů zařazených do primární analýzy úplné odpovědi.
{Rozdíl a interval spolehlivosti (CI) byly vypočítány pomocí metody, kterou nyvrhli Miettinen a Nurminen a byly upraveny podle pohlaví.
{Úplná odpověď = žádné zvracení a žádné použití záchranné terapie.
§Celkem = 0 až 120 hodin od zahájení chemoterapie cisplatinou.
§§Pozdní fáze = 25 až 120 hodin od zahájení chemoterapie cisplatinou.
Pediatrická populace
Studie hodnotící použití aprepitantu u pediatrických pacientů probíhají (ohledně informací o pediatrickém použití viz bod 4.2).
5.2 Farmakokinetické vlastnosti
Aprepitant vykazuje nelineární farmakokinetiku. Jak clearance, tak absolutní biologická dostupnost se snižují s rostoucí dávkou.
Absorpce
AUC0-<x, aprepitantu po perorálním podání dávky 165 mg byla ekvivalentní AUC0-OT po dávce 150 mg fosaprepitantu podané intravenózně, zatímco Cmax byla 2,4krát nižší.
Po jediné perorální dávce 165 mg aprepitantu zdravým dobrovolníkům byla střední hodnota AUC0-<X) aprepitantu 32,5 ^g»hod/ml a střední hodnota maximálních koncentrací aprepitantu byla 1,67 ^g/ml.
Střední hodnota maximálních plazmatických koncentrací (Cmax) aprepitantu nastala za přibližně 4 hodiny ((tmax). Perorální podání tobolky se standardní lehkou snídaní a silně tučnou snídaní vedlo k až 8%, respektive 47% zvýšení AUC0-<X) aprepitantu. Tento vzestup se nepovažuje za klinicky relevantní.
Distribuce
Aprepitant se ve vysoké míře váže na proteiny, se střední hodnotou 97 %. Geometrická střední hodnota zdánlivého distribučního objemu v rovnovážném stavu (Vdss) je u člověka přibližně 66 litrů.
Biotransformace
Aprepitant prochází rozsáhlou biotransformací. U zdravých mladých dospělých jedinců vykazuje aprepitant po dobu 72 hodin po jednorázové intravenózní aplikaci 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, v plazmě přibližně 19 % radioaktivity, což ukazuje na značnou přítomnost metabolitů v plazmě. V lidské plazmě bylo zjištěno dvanáct metabolitů aprepitantu. Metabolismus aprepitantu probíhá ve velké míře cestou oxidace na morfolinovém kruhu a jeho postranních řetězcích a výsledné metabolity jsou pouze slabě aktivní. In vitro studie na lidských jaterních mikrozómech ukázaly, že aprepitant se biotransformuje primárně cestou CYP3A4, případně s malým podílem CYP1A2 a CYP2C19.
Aprepitant se v nezměněné podobě močí nevylučuje. Metabolity se vylučují močí a žlučí ve stolici.
Po jednorázově intravenózně aplikované dávce 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, zdravým jedincům bylo 57 % radioaktivity zjištěno v moči a 45 % ve stolici.
Plazmatická clearance aprepitantu závisí na dávce, se zvyšující se dávkou se snižuje a v rozmezí terapeutických dávek se pohybovala přibližně na hodnotách od 60 do 72 ml/min. Terminální poločas se pohybuje od přibližně 9 do 13 hodin.
Farmakokinetika u speciálních populací
Starší osoby: po perorálním podání jedné 125mg dávky aprepitantu 1. den a dávky 80 mg jednou denně 2. až 5. den byla hodnota AUC0-24 hod aprepitantu 1. den o 21 % vyšší a 5. den o 36 % vyšší u starších jedinců (> 65 let) ve srovnání s mladšími dospělými. Hodnota Cmax byla u starších ve srovnání s mladšími dospělými 1. den o 10 % vyšší a 5. den o 24 % vyšší. Tyto rozdíly se nepovažují za klinicky významné. U starších pacientů není třeba dávkování přípravku EMEND nijak upravovat.
Pohlaví: po perorálním podání jediné 125mg dávky aprepitantu byla hodnota Cmax aprepitantu u žen ve srovnání s muži o 16 % vyšší. Poločas aprepitantu je u žen ve srovnání s muži o 25 % nižší a k dosažení tnax dochází zhruba ve stejnou dobu. Tyto rozdíly nejsou považovány za klinicky významné. Dávkování přípravku EMEND není nutno podle pohlaví pacienta nijak upravovat.
Porucha funkce jater: mírná porucha funkce jater (Childovo-Pughova třída A) farmakokinetiku aprepitantu v klinicky významné míře neovlivňuje. U pacientů s mírnou poruchou funkce jater není třeba dávkování nijak upravovat. Z dostupných dat nelze činit žádné závěry ohledně vlivu středně těžké poruchy funkce jater (Childovo-Pughova třída B) na farmakokinetiku aprepitantu. K dispozici nejsou žádné klinické ani farmakokinetické údaje o pacientech s těžkou poruchou funkce jater (Childovo-Pughova třída C).
Porucha funkce ledvin: pacientům s těžkou poruchou funkce ledvin (CrCl < 30 ml/min) a pacientům s terminálním renálním onemocněním (end stage renal disease, ESRD) s potřebou hemodialýzy byla podána jednorázová 240mg dávka aprepitantu.
U pacientů s těžkou poruchou funkce ledvin se hodnota AUC0_W celkového aprepitantu (nevázaného i vázaného na proteiny) ve srovnání se zdravými jedinci snížila o 21 % a hodnota Cmax se snížila o 32 %. U pacientů s ESRD podstupujících hemodialýzu se hodnota AUC0_<» celkového aprepitantu snížila o 42 % a hodnota Cmax se snížila o 32 %. Vzhledem k mírnému poklesu vazby aprepitantu na proteiny u pacientů s renálním onemocněním nebyla hodnota AUC farmakologicky aktivní části nenavázaného léku u pacientů s poruchou funkce ledvin ve srovnání se zdravými jedinci významně ovlivněna. Hemodialýza prováděná 4 nebo 48 hodin po podání dávky neměla na farmakokinetiku aprepitantu významný účinek; v dialyzátu bylo zjištěno méně než 0,2 % dávky.
U pacientů s poruchou funkce ledvin ani u pacientů s ESRD podstupujících hemodialýzu není zapotřebí dávkování přípravku EMEND nijak upravovat.
Vztah mezi koncentrací a účinkem
Studie využívající pozitronovou emisní tomografii (PET) s použitím vysoce specifického značení receptorů NK u zdravých mladých mužů ukázaly, že aprepitant prostupuje do mozku a obsazuje receptory NK v závislosti na dávce a koncentraci v plazmě. Předpokládá se, že plazmatické koncentrace aprepitantu dosažené při 3-denním léčebném režimu s přípravkem EMEND zajišťují více než 95% obsazení receptorů NK v mozku.
Pozitronová emisí tomografie u zdravých dobrovolníků mužů, kterým byla podána jediná perorální 165mg dávka aprepitant nebo jediná intravenózní 150mg dávka fosaprepitantu prokázala podobné obsazení receptorů NK v mozku při tmax, (> 99 %), za 24 hodin (> 99 %), 48 hodin (> 97 %) a 120 hodin (37 až 76 %) po podání dávky.
Obsazení receptorů NK1 v mozku aprepitantem dobře koreluje s plazmatickými koncentracemi aprepitantu.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické údaje získané na základě konvenčních studií toxicity, genotoxicity, hodnocení kancerogenního potenciálu, reprodukční a vývojové toxicity po jednorázovém a opakovaném podání přípravku neodhalily žádné zvláštní riziko pro člověka. Je nutno mít na paměti, že systémová expozice u hlodavců byla podobná nebo nižší, než terapeutická expozice u člověka při dávkách 125 mg/80 mg a 165 mg. Zejména je nutno zdůraznit, že i když v reprodukčních studiích nebyly při úrovních expozice odpovídajících lidským pozorovány žádné nežádoucí účinky na reprodukci, nepostačují expozice u zvířat k odpovídajícímu vyhodnocení rizika u člověka.
Ve studii juvenilní toxicity na potkanech, kterým se podával aprepitant od 10. do 63. postnatálního dne, docházelo u samic k časnému vaginálnímu otevírání od dávek 250 mg/kg dvakrát denně a u samců k opožděné prepuciální separaci, a to od dávek 10 mg/kg dvakrát denně. Nebyl zde žádný odstup od klinicky relevantní expozice. Nedošlo k žádným účinkům souvisejícím s léčbou ohledně páření, fertility či embryonálního/fetálního vývoje a nevyskytly se žádné patologické změny reprodukčních orgánů. Ve studii juvenilní toxicity na psech ošetřovaných od 14. do 42. postnatálního dne byla u samců při dávce 6 mg/kg/den pozorována snížená hmotnost varlat a velikost Leydigových buněk a u samic při dávkách od 4 mg/kg/den byla pozorována zvýšená hmotnost dělohy, hypertrofie dělohy a krčku a otok vaginálních tkání. Nebyl zde žádný odstup od klinicky relevantní expozice aprepitantu. Při krátkodobé léčbě podle doporučeného dávkovacího režimu se klinická relevance těchto zjištění nepovažuje za pravděpodobnou.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky Sacharóza
Mikrokrystalická celulóza (E 460)
Hyprolóza (E 463)
Natrium-lauryl-sulfát
Obal tobolky Želatina
Oxid titaničitý (E 171)
Indigokarmín (E 132)
Inkoust na potisk Šelak
Hydroxid draselný Černý oxid železitý (E 172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Hliníkový blistr obsahující jednu 165mg tobolku.
6 hliníkových blistrů, každý obsahující jednu 165mg tobolku.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO
EU/1/03/262/009
EU/1/03/262/010
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. listopadu 2003 Datum prodloužení registrace: 22. září 2008
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
EMEND 125 mg tvrdé tobolky EMEND 80 mg tvrdé tobolky
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna 125mg tobolka obsahuje aprepitantum 125 mg. Jedna 80mg tobolka obsahuje aprepitantum 80 mg.
Pomocná látka se známým účinkem
Jedna tobolka obsahuje 125 mg sacharózy (v 125 mg tvrdé tobolce).
Pomocná látka se známým účinkem
Jedna tobolka obsahuje 80 mg sacharózy (v 80 mg tvrdé tobolce).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Tvrdá tobolka.
125mg tobolky jsou matné s bílým tělem a růžovým víčkem; na těle je cirkulárně černým inkoustem vytištěno „462“ a „125 mg“. 80mg tobolky jsou matné s bílým tělem a víčkem; na těle je cikulárně černým inkoustem vytištěno „461“ a „80 mg“.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence nauzey a zvracení spojených s vysoce a středně silně emetogenní protinádorovou chemoterapií u dospělých a dospívajících od 12 let věku.
EMEND 125 mg/80 mg se podává jako součást kombinované terapie (viz bod 4.2).
4.2 Dávkování a způsob podání
Dávkování
Dospělí
EMEND se podává po dobu 3 dní jako součást léčebného režimu zahrnujícího kortikosteroid a antagonistu 5-HT3. Doporučená dávka je 125 mg perorálně jednou denně jednu hodinu před zahájením chemoterapie 1. den a 80 mg perorálně jednou denně 2. a 3. den ráno.
K prevenci nevolnosti a zvracení v souvislosti s emetogenní protinádorovou chemoterapií se u dospělých doporučují následující režimy:
Vysoce emetogenní chemoterapeutický režim
|
1. den |
2. den |
3. den |
4. den | |
|
EMEND |
125 mg perorálně |
80 mg perorálně |
80 mg perorálně |
žádný |
|
Dexamethason |
12 mg perorálně |
8 mg perorálně |
8 mg perorálně |
8 mg perorálně |
|
Antagonisté 5- |
Standardní dávka |
žádný |
žádný |
žádný |
|
HT3 |
antagonistů 5-HT3. | |||
|
Ohledně příslušného dávkování zvoleného | ||||
|
antagonisty 5-HT3 viz informace o daném přípravku |
Dexamethason se musí podávat 1. den 30 minut před zahájením chemoterapie a 2. až 4. den ráno. Dávka dexamethasonu přispívá k interakcím léčivých látek.
|
Středně emetogenní chemoterapeutický režim 1. den |
2. den |
3. den | |
|
EMEND |
125 mg perorálně |
80 mg perorálně |
80 mg perorálně |
|
Dexamethason |
12 mg perorálně |
žádný |
žádný |
|
Antagonisté 5-HT3 |
Standardní dávka antagonistů 5-HT3. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném |
žádný |
žádný |
přípravku
Dexamethason se musí podávat 1. den 30 minut před zahájením chemoterapie. Dávka dexamethasonu přispívá k interakcím léčivých látek.
Pediatrická populace Dospívající (ve věku 12 až 17 let)
Přípravek EMEND se podává 3 dny jako součást režimu, který zahrnuje antagonistu 5-HT3. Doporučené dávkování tobolek přípravku EMEND je 125 mg perorálně 1. den a 80 mg perorálně 2. a 3. den. Přípravek EMEND se podává perorálně 1 hodinu před chemoterapií 1., 2. a 3. den. Pokud se 2. a 3. den nepodává žádná chemoterapie, má být přípravek EMEND podáván ráno. Ohledně správného dávkování zvoleného antagonisty 5-HT3 nahlédněte do jeho souhrnu údajů o přípravku (SPC). Pokud se s přípravkem EMEND podává současně kortikosteroid, jako je dexamethason, má se podat 50 % obvyklé dávky kortikosteroidu (viz body 4.5 a 5.1).
Bezpečnost a účinnost 80mg a 125mg tobolky nebyla u dětí mladších 12 let prokázána. K dispozici nejsou žádné údaje. Viz SPC prášku pro perorální suspenzi pokud jde o vhodné dávkování u kojenců, batolat a dětí ve věku od 6 měsíců do méně než 12 let.
Obecně
Množství údajů o účinnosti při kombinaci s jinými kortikosteroidy a antagonisty 5-HT3 je omezeno. Další informace o současném podávání s kortikosteroidy jsou uvedeny v bodu 4.5. U současně podávaných léčivých přípravků obsahujících antagonistu 5-HT3 nahlédněte do jejich SPC.
Zvláštní _ populace Starší osoby (> 65 let)
U starších osob není nutno dávku nijak upravovat (viz bod 5.2).
Pohlaví
S ohledem na pohlaví není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin ani u pacientů v terminálním stádiu renálního onemocnění podstupujících hemodialýzu není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou poruchou funkce jater není nutno dávku nijak upravovat. Pokud se týče pacientů se středně těžkou poruchou funkce jater, je k dispozici pouze omezené množství dat a nejsou dostupné žádné údaje od pacientů s těžkou poruchou funkce jater. Těmto pacientům se aprepitant musí podávat opatrně (viz body 4.4 a 5.2).
Způsob podání
Tvrdou tobolku je nutno polknout vcelku.
Přípravek EMEND lze užívat bez ohledu na jídlo.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Současné podávání s pimozidem, terfenadinem, astemizolem nebo cisapridem (viz bod 4.5).
4.4 Zvláštní upozornění a opatření pro použití Pacienti se středně těžkou až těžkou poruchou funkce jater
Pro pacienty se středně těžkou poruchou funkce jater existuje pouze omezené množství dat a žádné údaje nejsou k dispozici pro pacienty s těžkou poruchou funkce jater. U těchto pacientů je nutno přípravek EMEND používat s opatrností (viz bod 5.2).
Interakce na CYP3A4
EMEND je nutno podávat s opatrností pacientům současně užívajícím perorálně podávané léčivé látky, které se primárně metabolizují cestou CYP3A4 a které mají úzké terapeutické rozmezí, jako je cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, deriváty námelových alkaloidů, fentanyl a chinidin (viz bod 4.5). Navíc je nutno k podávání současně s irinotekanem přistupovat s obzvláštní opatrností, protože uvedená kombinace by mohla mít za následek zvýšenou toxicitu.
Současné podávání s warfarinem (substrát CYP2C9)
U pacientů dlouhodobě léčených warfarinem je nutno hodnotu mezinárodního normalizovaného poměru (INR) pozorně sledovat během léčby přípravkem EMEND a po dobu 14 dnů po každé 3-denní léčbě přípravkem EMEND (viz bod 4.5).
Současné podávání s hormonální antikoncepcí
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a ještě 2 měsíce po poslední dávce přípravku EMEND je třeba používat alternativní nehormonální záložní antikoncepční metody (viz bod 4.5).
Pomocné látky
Tobolky přípravku EMEND obsahují sacharózu. Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy, malabsorpcí glukózy-galaktózy nebo deficitem sacharáza-izomaltázy nemají tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aprepitant (125 mg/80 mg) je substrát, středně silný inhibitor a induktor CYP3A4. Aprepitant je také induktor CYP2C9. Během léčby přípravkem EMEND je inhibován CYP3A4. Po ukončení léčby přípravkem EMEND dochází k přechodné mírné indukci CYP2C9, CYP3A4 a glukuronidace. Nezdá se, že by aprepitant interagoval s P glykoproteinovým transportérem, jak naznačuje nepřítomnost interakce aprepitantu s digoxinem.
Účinek aprepitantu na farmakokinetiku dalších léčivých látek Inhibice CYP3A4
Jako středně silný inhibitor CYP3A4 může aprepitant (125 mg/80 mg) zvýšit plazmatické koncentrace současně podávaných léčivých látek, které se metabolizují cestou CYP3A4. Celková expozice perorálně podávaným substrátům CYP3A4 se může zvýšit přibližně 3krát během 3denní léčby přípravkem EMEND; předpokládá se, že účinek aprepitantu na plazmatické koncentrace intravenózně podávaných substrátů CYP3A4 bude menší. Přípravek EMEND se nesmí podávat současně s pimozidem, terfenadinem, astemizolem nebo cisapridem (viz bod 4.3). Inhibice CYP3A4 aprepitantem by mohla mít za následek zvýšené plazmatické koncentrace uvedených léčivých látek, což může vyvolat závažné nebo život ohrožující reakce. Při současném podávání přípravku EMEND a perorálně podávaných léčivých látek, které jsou metabolizovány převážně prostřednictvím CYP3A4 a které mají úzké terapeutické rozmezí, jako je cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, diergotamin, ergotamin, fentanyl a chinidin, se doporučuje opatrnost (viz bod 4.4).
Kortikosteroidy
Dexamethason: Obvyklou dávku perorálně podaného dexamethasonu je nutno při současném podávání s přípravkem EMEND v režimu 125 mg/80 mg snížit přibližně o 50 %. Dávka dexamethasonu v klinických studiích nevolnosti a zvracení vyvolaných chemoterapií byla vybrána s ohledem na interakce léčivých látek (viz bod 4.2). Pokud se EMEND podával v léčebném režimu v dávce 125 mg spolu s dexamethasonem 20 mg perorálně první den, a poté v dávce 80 mg/den spolu s dexamethasonem 8 mg perorálně druhý až pátý den, došlo první a pátý den k 2,2-násobnému zvýšení hodnoty AUC dexamethasonu, který je substrátem CYP3A4.
Methylprednisolon: Při současném podávání s přípravkem EMEND v režimu 125 mg/80 mg je třeba obvyklou dávku intravenózně aplikovaného methylprednisolonu snížit přibližně o 25 %, a obvyklou dávku perorálně podávaného methylprednisolonu přibližně o 50 %. Pokud se EMEND podával v léčebném režimu v dávce 125 mg první den a 80 mg/den 2. a 3. den, zvýšil hodnotu AUC methylprednisolonu, který je substrátem CYP3A4, 1. den 1,3krát a 3. den 2,5krát v případě, že se methylprednisolon podával současně intravenózně v dávce 125 mg první den a perorálně v dávce 40 mg 2. a 3. den.
V kontinuální léčbě methylprednisolonem se může hodnota AUC methylprednisolonu koncem 2-týdenního období po zahájení podávání přípravku EMEND snížit v důsledku indukčního účinku aprepitantu na CYP3A4. Lze očekávat, že tento účinek bude u perorálně podávaného methylprednisolonu výraznější.
Chemoterapeutické léčivé přípravky
Ve farmakokinetických studiích neměl EMEND při podávání v režimu 125 mg 1. den a 80 mg/den 2. a 3. den, vliv na farmakokinetiku docetaxelu podaného intravenózně 1. den nebo vinorelbinu podaného intravenózně 1. nebo 8. den. Protože účinek přípravku EMEND na farmakokinetiku perorálně podávaných substrátů CYP3A4 je větší než účinek přípravku EMEND na farmakokinetiku intravenózně podávaných substrátů CYP3A4, nelze vyloučit interakce s perorálně podávanými chemoterapeutickými léčivými přípravky, které se biotransformují převážně nebo částečně cestou CYP3A4 (např. etoposid, vinorelbin). U pacientů dostávajících léčivé přípravky, které se metabolizují především nebo i částečně CYP3A4, se doporučuje opatrnost, přičemž může být vhodné tyto pacienty dodatečně sledovat (viz bod 4.4). Po uvedení na trh byly hlášeny při současném podávání aprepitantu a ifosfamidu případy neurotoxicity, potenciální nežádoucí účinek ifosfamidu.
Imunosupresiva
V průběhu 3denního režimu léčby nevolnosti a zvracení vyvolaných chemoterapií se předpokládá přechodné střední zvýšení následované mírným poklesem expozice imunosupresivům metabolizovaným prostřednictvím CYP3A4 (např. cyklosporin, takrolimus, everolimus a sirolimus).
S ohledem na krátké trvání 3denního režimu a na časově závislé omezené změny expozice se během 3 dnů současného podávání s přípravkem EMEND snížení dávky imunosupresiv nedoporučuje.
Midazolam
Při současném podávání midazolamu nebo jiných benzodiazepinů metabolizovaných cestou CYP3A4 (alprazolam, triazolam) spolu s přípravkem EMEND (125 mg/80 mg) je nutno vzít v úvahu možné účinky zvýšených plazmatických koncentrací těchto léčivých přípravků.
EMEND zvýšil hodnotu AUC midazolamu, který je citlivým substrátem CYP3A4, 2,3krát 1. den a 3,3krát 5. den, pokud byl midazolam podán perorálně v jediné 2mg dávce 1. a 5. den léčebného režimu s přípravkem EMEND v dávce 125 mg 1. den a 80 mg/den 2. až 5. den.
V jiné studii s intravenózní aplikací midazolamu se EMEND podával v dávce 125 mg 1. den a 80 mg/den 2. a 3. den a midazolam v dávce 2 mg se aplikoval intravenózně před zahájením 3denního léčebného režimu s přípravkem EMEND a 4., 8., a 15. den. EMEND zvýšil hodnotu AUC midazolamu o 25 % 4. den a snížil hodnotu AUC midazolamu o 19 % 8. den a o 4 % 15. den. Tyto účinky nebyly považovány za klinicky významné.
Ve třetí studii s intravenózní a perorální aplikací midazolamu se EMEND podával v dávce 125 mg 1. den a 80 mg/den 2. a 3. den společně s ondansetronem 32 mg 1. den, dexamethasonem 12 mg 1. den a 8 mg 2. až 4. den. Tato kombinace (tedy EMEND, ondansetron a dexamethason) snížila hodnotu AUC perorálního midazolamu o 16 % 6. den, o 9 % 8. den, o 7 % 15. den a o 17 % 22. den. Tyto účinky nebyly považovány za klinicky významné.
Byla provedena dodatečná studie intravenózního podávání midazolamu a přípravku EMEND. Midazolam v dávce 2 mg byl podán intravenózně 1 hodinu po perorálně podané jedné 125mg dávce přípravku EMEND. Plazmatická hodnota AUC midazolamu se zvýšila 1,5krát. Tento účinek nebyl považován za klinicky významný.
Indukce
Jako mírný induktor CYP2C9, CYP3A4 a glukuronidace může aprepitant během dvou týdnů po zahájení a léčbě snižovat plazmatické koncentrace substrátů eliminovaných těmito cestami. Tento účinek se může projevit až po skončení třídenní léčby přípravkem EMEND. U substrátů CYP2C9 a CYP3A4 je indukce přechodná s dosažením maxima účinku 3 až 5 dní po skončení třídenní léčebné kúry přípravkem EMEND. Účinek se zachovává po dobu několika dní, poté se zvolna ztrácí a dva týdny po skončení léčby přípravkem EMEND je již klinicky nevýznamný. Mírnou indukci glukuronidace lze pozorovat i při perorálním podávání aprepitantu v dávce 80 mg po dobu 7 dní.
Údaje ohledně účinku na CYP2C8 a CYP2C19 nejsou k dispozici. Opatrnost se doporučuje v případech, kdy se v daném období podávají warfarin, acenokumarol, tolbutamid, fenytoin nebo jiné léčivé látky, o nichž je známo, že jsou metabolizovány CYP2C9.
Warfarin
U pacientů dlouhodobě léčených warfarinem je třeba pozorně sledovat protrombinový čas (INR) během léčby přípravkem EMEND a po dobu 2 týdnů po každé 3denní léčbě přípravkem EMEND z důvodu chemoterapií vyvolané nevolnosti a zvracení (viz bod 4.4). Jestliže se podala jediná 125mg dávka přípravku EMEND 1. den a dávka 80 mg/den 2. a 3. den zdravým jedincům ve stabilizovaném stavu při dlouhodobé léčbě warfarinem, neměl EMEND 3. den žádný účinek na hodnotu plazmatické AUC pro R(+) ani S(-) warfarin, nicméně 5 dní po skončení podávání přípravku EMEND, ale došlo ke 34% snížení dolní hodnoty koncentrace S(-) warfarinu (substrátu CYP2C9) spolu se 14% poklesem hodnoty INR.
Tolbutamid
Pokud se EMEND podával v dávce 125 mg 1. den a v dávce 80 mg/den 2. a 3. den, snížil 4. den hodnotu AUC tolbutamidu (substrátu CYP2C9) o 23 %, 8. den o 28 %, a 15. den o 15 % v případě, že se jediná 500mg dávka tolbutamidu podala perorálně před zahájením 3denní kúry s přípravkem EMEND a pak 4., 8., a 15. den.
Hormonální antikoncepce
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a ještě 2 měsíce po poslední dávce přípravku EMEND je třeba používat alternativní nehormonální záložní antikoncepční metody.
V klinické studii byly 1. až 21. den podávány jednorázové dávky perorální antikoncepce obsahující ethinylestradiol a norethisteron spolu s přípravkem EMEND, podávaným v režimu v dávce 125 mg 8. den a 80 mg/den 9. a 10. den s ondansetronem v dávce 32 mg podaným intravenózně 8. den
a dexamethasonem v dávce 12 mg podaným perorálně 8. den a dále 8 mg/den 9., 10. a 11. den. V této studii se minimální koncentrace ethinylestradiolu snížily 9. až 21. den až o 64 % a minimální koncentrace norethisteronu až o 60 %.
Antagonisté 5-HT3
V klinických studiích lékových interakcí nevykazoval aprepitant žádné klinicky významné účinky na farmakokinetiku ondansetronu, granisetronu nebo hydrodolasetronu (aktivní metabolit dolasetronu).
Účinky jiných léčivých přípravků na farmakokinetiku aprepitantu
K současnému podávání přípravku EMEND s léčivými látkami, které inhibují aktivitu CYP3A4 (např. ketokonazol, itrakonazol, vorikonazol, posakonazol, klarithromycin, telithromycin, nefazodon a inhibitory proteázy), je třeba přistupovat opatrně, protože se předpokládá, že tato kombinace povede k několikanásobnému zvýšení plazmatické koncentrace aprepitantu (viz bod 4.4).
Je nutno se vyvarovat současného podávání přípravku EMEND s léčivými látkami, které silně indukují aktivitu CYP3A4 (např. s rifampicinem, fenytoinem, karbamazepinem, fenobarbitalem), protože tato kombinace vede ke snížení plazmatických koncentrací aprepitantu, což může vést ke snížení účinnosti přípravku EMEND. Současné podávání přípravku EMEND s bylinnými přípravky obsahujícími třezalku tečkovanou (Hypericumperforatum) se nedoporučuje.
Ketokonazol
Jestliže se podal aprepitant v jediné 125mg dávce 5. den 10denního léčebného režimu se silným inhibitorem ketokonazolem podaným v dávce 400 mg/den, zvýšila se hodnota AUC aprepitantu přibližně 5krát a střední hodnota terminálního poločasu aprepitantu se zvýšila přibližně 3krát.
Rifampicin
Jestliže se podal aprepitant v jediné 375 mg dávce 9. den 14denního léčebného režimu se silným induktorem rifampicinem podaným v dávce 600 mg/den, snížila se hodnota AUC aprepitantu o 91 % a střední hodnota terminálního poločasu aprepitantu se snížila o 68 %.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a 2 měsíce po poslední dávce přípravku EMEND je nutno používat alternativní nehormonální záložní antikoncepční metody (viz body 4.4 a 4.5).
Nejsou dostupné žádné klinické údaje týkající se těhotenství vystavených aprepitantu. Potenciální reprodukční toxicita aprepitantu nebylazcela stanovena, protože míry expozice vyšší než hodnoty terapeutické expozice u člověka při dávce 125 mg/80 mg nebylo možno ve studiích se zvířaty dosáhnout. Tyto studie nenaznačily žádné přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Možné účinky na reprodukci změn v regulaci neurokininů nejsou známy. Přípravek EMEND by neměl být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Aprepitant se vylučuje do mléka kojících potkanů. Není známo, zda se aprepitant vylučuje do lidského mateřského mléka, a proto se kojení během léčby přípravkem EMEND nedoporučuje.
Fertilita
Potenciál aprepitantu navodit účinky na fertilitu nebyl dosud plně popsán, protože expozičních hladin přesahujících terapeutickou expozici u lidí nemohlo být ve studiích na zvířatech dosaženo. Tyto studie fertility neukazují na přímé nebo nepřímé škodlivé účinky ohledně páření, fertility, embryonálního/fetálního vývoje či počtu a motility spermií (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek EMEND může mít mírný vliv na schopnost řídit, jezdit na kole nebo obsluhovat stroje. Po užití přípravku EMEND se mohou objevit závratě a únava (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Profil bezpečnosti aprepitantu byl hodnocen přibližně u 6 500 dospělých ve více než 50 studiích a 184 dětí a dospívajících ve 2 kontrolovaných pediatrických klinických studiích.
Nejčastějšími nežádoucími účinky, uváděnými ve vyšším výskytu u dospělých léčených režimem s aprepitantem než standardní terapií, u pacientů léčených vysoce emetogenní chemoterapií byly: škytavka (4,6 % oproti 2,9 %), zvýšení ALT (2,8 %, oproti 1,1 %), dyspepsie (2,6 %, oproti 2,0 %), zácpa (2,4 %, oproti 2,0 %), bolest hlavy (2,0 %, oproti 1,8 %) a snížení chuti k jídlu (2,0 %, oproti 0,5 %). Nejčastějším nežádoucím účinkem uváděným ve vyšším výskytu u pacientů léčených režimem s aprepitantem než standardní terapií u pacientů léčených středně emetogenní chemoterapií byla únava (1,4 %, oproti 0,9 %).
Nejčastějšími nežádoucími účinky hlášenými s vyšší incidencí u pediatrických pacientů léčených režimem aprepitantu než u kontrolního režimu během léčby emetogenní protinádorovou chemoterapií byla škytavka (3,3 % versus 0,0 %) a zarudnutí (1,1 % versus 0,0 %).
Tabulkový seznam nežádoucích účinků
Následující nežádoucí účinky byly pozorovány v souhrnné analýze studií s vysoce emetogenní chemoterapií a středně emetogenní chemoterapií u pacientů léčených režimem s aprepitantem s vyšší incidencí než při standardní terapii dospělých nebo pediatrických pacientů nebo po uvedení přípravku na trh. Kategorie frekvence uvedené v tabulce jsou založeny na studiích u dospělých; pozorované frekvence v pediatrických studiích byly podobné nebo nižší, není-li v tabulce uvedeno. Některé méně časté nežádoucí účinky v dospělé populaci nebyly pozorovány v pediatrických studiích.
Frekvence jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000) a velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Frekvence |
|
Infekce a infestace |
kandidóza, stafylokokové infekce |
vzácné |
|
Poruchy krve a lymfatického systému |
febrilní neutropenie, anemie |
méně časté |
|
Poruchy imunitního systému |
hypersenzitivní reakce včetně anafylaktických reakcí |
není známo |
|
Poruchy metabolismu a výživy |
snížení chuti k jídlu |
časté |
|
polydipsie |
vzácné | |
|
Psychiatrické poruchy |
méně časté | |
|
dezorientace, euforická nálada |
vzácné | |
|
Poruchy nervového systému |
časté | |
|
závrať, somnolence |
méně časté |
|
Třída orgánových systémů |
Nežádoucí účinek |
Frekvence |
|
kognitivní poruchy, letargie, poruchy vnímání chutí |
vzácné | |
|
Poruchy oka |
konjunktivitida |
vzácné |
|
Poruchy ucha a labyrintu |
tinnitus |
vzácné |
|
Srdeční poruchy |
palpitace |
méně časté |
|
bradykardie, kardiovaskulární porucha |
vzácné | |
|
Cévní poruchy |
návaly horka/zarudnutí |
méně časté |
|
Respirační, hrudní a mediastinální poruchy |
škytavka |
časté |
|
bolest v orofaryngu, kýchání, kašel, postnazální zatékání hlenu (postnasal drip), podráždění hrdla |
vzácné | |
|
Gastrointestinální poruchy |
zácpa, dyspepsie |
časté |
|
říhání, nauzeaT, zvraceníT, gastroesofageální refluxní choroba, bolest břicha, sucho v ústech, flatulence |
méně časté | |
|
perforace duodenálního vředu, stomatitida, abdominální distenze, tvrdá stolice, neutropenická kolitida |
vzácné | |
|
Poruchy kůže a podkožní tkáně |
vyrážka, akné |
méně časté |
|
fotosenzitivní reakce, hyperhidróza, seborea, kožní léze, svědivá vyrážka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza |
vzácné | |
|
svědění, kopřivka |
není známo | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
svalová slabost, svalové spasmy |
vzácné |
|
Poruchy ledvin a močových cest |
méně časté | |
|
polakisurie |
vzácné | |
|
Celkové poruchy a reakce v místě aplikace |
časté | |
|
astenie, celkový pocit nemoci |
méně časté | |
|
edém, pocit nepohody na hrudi, poruchy chůze |
vzácné | |
|
Vyšetření |
zvýšení ALT |
časté |
|
zvýšení AST, zvýšení alkalické fosfatázy |
méně časté | |
|
pozitivní test na červené krvinky v moči, zvýšení hladiny sodíku v krvi, pokles tělesné hmostnosti, pokles počtu neutrofilů, přítomnost glukózy v moči, zvýšená tvorba moči |
vzácné |
fNausea a zvracení byly parametry účinnosti v prvních 5-ti dnech následujících po chemoterapeutické léčbě a byly hlášeny jako nežádoucí příhody pouze poté.
Popis vybraných nežádoucích účinků
Profily nežádoucích reakcí u dospělých v prodloužených studiích s vysoce nebo středně emetogenní chemoterapií s opakovanými až 6 dalšími cykly chemoterapie byly celkově podobné jako profily pozorované v 1. cyklu.
V další aktivním komparátorem kontrolované klinické studii u 1 169 dospělých pacientů léčených aprepitantem a vysoce emetogenní terapií byl profil nežádoucích účinků obecně podobný profilu, který byl pozorovaný v ostatních studiích s vysoce emetogenní terapií s aprepitantem.
U dospělých pacientů léčených aprepitantem pro pooperační nevolnost a zvracení (PONV) byly pozorovány další nežádoucí reakce s větším výskytem než u ondansetronu: bolest horní poloviny břicha, abnormální střevní zvuky, zácpa*, dysartrie, dušnost, hypoestézie, insomnie, mióza, nevolnost, smyslové poruchy, žaludeční dyskomfort, subileus*, snížená ostrost vidění, sípot.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování je nutno EMEND vysadit a zajistit obecnou podpůrnou léčbu a sledování pacienta. Vzhledem k antiemetickému účinku aprepitantu může snaha o vyvolání zvracení pomocí léčivých přípravků selhat.
Aprepitant nelze odstranit hemodialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiemetika a antinauzeancia, ATC kód: A04AD12
Aprepitant je selektivní antagonista s vysokou afinitou pro receptory neurokininu 1 (NKi) lidské substance P.
Třídenní režim aprepitantu u dospělých
Ve 2 randomizovaných, dvojitě zaslepených studiích zahrnujících celkem 1 094 dospělých léčených chemoterapií obsahující cisplatinu > 70 mg/m2, byl aprepitant v kombinaci s léčebným režimem ondansetron/dexamethason (viz bod 4.2) srovnáván se standardním režimem (placebo plus ondansetron 32 mg aplikovaný intravenózně 1. den plus dexamethason 20 mg perorálně podaný 1. den a 8 mg perorálně dvakrát denně 2. až 4. den). I když se v klinických hodnoceních používala 32mg intravenózní dávka ondansetronu, nejedná se již o doporučenou dávku. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku.
Účinnost se hodnotila pomocí následujícího souhrnného parametru: úplná odpověď (definovaná jako žádné emetické epizody, bez použití „záchranné“ terapie) primárně v 1. cyklu. Výsledky se hodnotily v každé studii jednotlivě a pak pro tyto 2 studie dohromady.
Přehled hlavních výsledků studií z kombinované analýzy je uveden v tabulce 1.
Tabulka 1
Procenta dospělých pacientů léčených vysoce emetogenní chemoterapií s odpovědí na léčbu podle
|
léčebných skupin a fáze |
- 1. cyklus | ||
|
SOUHRNNÝ |
Režim |
Standardní |
Rozdíly* |
|
PARAMETR |
s aprepitantem |
terapie | |
|
(N = 521) ^ |
(N = 524)T |
% (95% CI) | |
|
% |
% |
Úplná odpověď (bez zvracení a „záchranné“ terapie)
|
Celkem (0-120 hodin) 0-24 hodin 25-120 hodin |
67,7 86,0 71,5 |
47,8 73.2 51.2 |
19,9 12,7 20,3 |
(14,0, 25,8) (7,9, 17,6) (14,5, 26,1) |
|
JEDNOTLIVÁ KRITÉRIA | ||||
|
Bez zvracení (žádné emetické epizody bez ohledu |
na použití „záchranné“ terapie) | |||
|
Celkem (0-120 hodin) |
71,9 |
49,7 |
22,2 |
(16,4, 28,0) |
|
0-24 hodin |
86,8 |
74,0 |
12,7 |
(8,0, 17,5) |
|
25-120 hodin |
76,2 |
53,5 |
22,6 |
(17,0, 28,2) |
|
Bez významné nevolnosti (maximálně VAS <25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
72,1 |
64,9 |
7,2 |
(1,6, 12,8) |
|
25-120 hodin |
74,0 |
66,9 |
7,1 |
(1,5, 12,6) |
* Intervaly spolehlivosti (CI) byly vypočítány bez korekce pro pohlaví a současnou chemoterapii, které byly zahrnuty do primární analýzy míry pravděpodobnosti a logistických modelů. t Jeden pacient v režimu pouze s aprepitantem měl data v akutní fázi a byl vyřazen z celkové a prodloužené fáze analýzy: jeden pacient pouze ve standardním režimu měl pouze data v prodloužené fázi a byl vyřazen z celkové a akutní fáze analýzy.
Odhad doby první epizody zvracení v kombinované analýze je zobrazen vynesením hodnot metodou Kaplana-Meiera v grafu 1.
Graf 1
Procento dospělých pacientů léčených vysoce emetogenní chemoterapií, u nichž nedošlo k epizodě zvracení - 1. cyklus
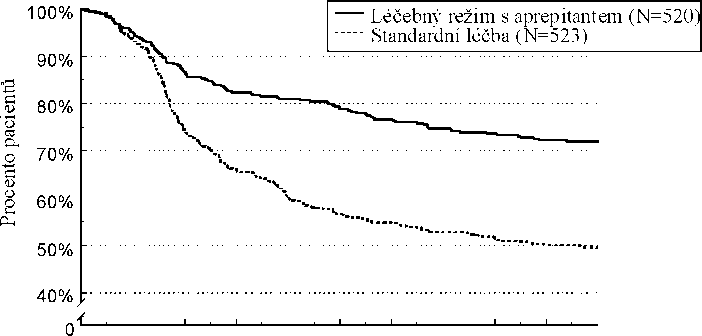
0 12 24 36 48 60 72 84 96 108 120
Čas (v hodinách)
Statisticky významné rozdíly v účinnosti byly pozorovány v každé z těchto 2 jednotlivých studií.
Ve těchto stejných 2 klinických studiích pokračovalo 851 dospělých pacientů v prodloužené studii s opakovanými až 5 dalšími cykly chemoterapie. Účinnost režimu s aprepitantem zůstala během všech cyklů zřejmě zachována.
V randomizované, dvojitě zaslepené studii s celkovým počtem 866 dospělých pacientů (864 žen,
2 muži) léčených chemoterapií, která zahrnovala cyklofosfamid 750-1 500 mg/m2; nebo cyklofosfamid 500-1 500 mg/m2 a doxorubicin (< 60 mg/m2) nebo epirubicin (< 100 mg/m2), byl aprepitant v kombinaci s režimem obsahujícím ondansetron/dexamethason (viz bod 4.2) srovnáván se standardní terapií (placebo plus ondansetron 8 mg perorálně (dvakrát 1. den a každých 12 hodin 2. a 3. den) plus dexamethason 20 mg perorálně 1. den).
Účinnost se hodnotila pomocí souhrnného parametru: úplná odpověď (definovaná jako žádné emetické epizody a nepoužití záchranné terapie) primárně během 1. cyklu.
Přehled hlavních výsledků studie je uveden v Tabulce 2.
Tabulka 2
Procenta dospělých pacientů odpovídajících na léčbu podle léčebné skupiny a fáze - 1. cyklus
|
středně emetogenní chemoterapie | |||
|
SOUHRNNÝ |
Režim |
Standardní |
Rozdíly* |
|
PARAMETR |
s aprepitante |
terapie | |
|
m (N = 433)t % |
(N = 424) % |
% (95 % CI) | |
|
Úplná odpověď (bez zvracení a |
„záchranné“ terapie) | |||
|
Celkem (0-120 hodin) |
50,8 |
42,5 |
8,3 |
(1,6, 15,0) |
|
0-24 hodin |
75,7 |
69,0 |
6,7 |
(0,7, 12,7) |
|
25-120 hodin |
55,4 |
49,1 |
6,3 |
(-0,4, 13,0) |
|
jednotlivá kritéria | ||||
|
Bez zvracení (žádné emetické epizody bez ohledu na |
použití „záchranné“ |
terapie) | ||
|
Celkem (0-120 hodin) |
75,7 |
58,7 |
17,0 |
(10,8, 23,2) |
|
0-24 hodin |
87,5 |
77,3 |
10,2 |
(5,1, 15,3) |
|
25-120 hodin |
80,8 |
69,1 |
11,7 |
(5,9, 17,5) |
|
Bez významné nevolnosti (maximálně VAS <25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
60,9 |
55,7 |
5,3 |
(-1,3, 11,9) |
|
0-24 hodin |
79,5 |
78,3 |
1,3 |
(-4,2, 6,8) |
|
25-120 hodin |
65,3 |
61,5 |
3,9 |
(-2,6, 10,3) |
* Intervaly spolehlivosti byly vypočítány bez korekce na věkovou kategorii (<55 let, >55 let) a skupiny hodnotících lékařů, které byly zahrnuty do primární analýzy míry pravděpodobnosti a logistických modelů.
t Jeden pacient v režimu pouze s aprepitantem měl data v akutní fázi a byl vyřazen z celkové a prodloužené fáze analýzy.
Ve stejné klinické studii pokračovalo 744 dospělých pacientů v prodloužené studii s opakovanými až 3 dalšími cykly chemoterapie. Účinnost režimu s aprepitantem zůstala během všech cyklů zřejmě zachována.
Ve druhé multicentrické, randomizované, dvojitě zaslepené studii s paralelní skupinou byl režim s aprepitantem porovnáván se standardní léčbou u 848 dospělých pacientů (652 žen, 196 mužů), kterým byl podáván chemoterapeutický režim, který zahrnoval jakékoli intravenózní dávky oxaliplatiny, karboplatiny, epirubicinu, idarubicinu, ifosfamidu, irinotekanu, daunorubicinu, doxorubicinu; cyklofosfamid intravenózně (< 1 500 mg/m2) nebo cytarabin intravenózně (>1 g/m2). Pacientům, kterým byl podáván režim s aprepitantem, byla chemoterapie podávána na různé typy nádorů, včetně 52 % s rakovinou prsu, 21 % s gastrointestinálními rakovinami včetně kolorektálního karcinomu, 13 % s rakovinou plic a 6 % s gynekologickými rakovinami. Režim s aprepitantem v kombinaci s režimem ondansetron/dexamethason (viz bod 4.2) byl porovnáván se standardní léčbou
(placebo v kombinaci s ondansetronem 8 mg perorálně (dvakrát 1. den a každých 12 hodin 2. a 3. den) plus dexamethason 20 mg perorálně 1. den).
Účinnost byla založena na vyhodnocení následujících primárních a klíčových sekundárních cílových parametrů účinnosti: žádné zvracení za celou dobu (0 až 120 hodin po chemoterapii), vyhodnocení bezpečnosti a snášenlivosti režimu s aprepitantem při chemoterapií vyvolané nevolnosti a zvracení (CINV) a úplná odpověď (definovaná jako žádné zvracení a žádné použití záchranné léčby) za celou dobu (0 až 120 hodin po chemoterapii). Navíc byla sledována jako exploratorní cílový parametr účinnosti, „Žádná významná nauzea“ (No Significant Nausea) za celou dobu (0 až 120 hodin po chemoterapii), jak v akutní, tak i prodloužené fázi formou post-hoc analýzy.
Souhrn klíčových výsledků studie je uveden v Tabulce 3.
Tabulka 3
Procento dospělých pacientů odpovídajících na léčbu podle léčebné skupiny a fáze pro studii 2 -
cyklus 1
středně emetogenní chemoterapie
Režim Standardní Rozdíly*
s aprepi- léčba
tantem (N = 406) % (95% CI)
(N = 425) %
%
Úplná odpověď (žádné zvracení a žádná záchranná léčba)
|
Celkem (0-120 hodin) 0-24 hodin 25-120 hodin |
68.7 89,2 70.8 |
56.3 80.3 60,9 |
12,4 8.9 9.9 |
(5,9, 18,9) (4,0, 13,8) (3,5, 16,3) |
|
Žádná emeze (žádné emetické epizody bez ohledu |
na použití záchranné léčby) | |||
|
Celkem (0-120 hodin) |
76,2 |
62,1 |
14,1 |
(7,9, 20,3) |
|
0-24 hodin |
92,0 |
83,7 |
8,3 |
(3,9, 12,7) |
|
25-120 hodin |
77,9 |
66,8 |
11,1 |
(5,1, 17,1) |
|
Žádná významná nauzea (maximální VAS <25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
73,6 |
66,4 |
7,2 |
(1,0, 13,4) |
|
0-24 hodin |
90,9 |
86,3 |
4,6 |
(0,2, 9,0) |
|
25-120 hodin |
74,9 |
69,5 |
5,4 |
(-0,7, 11,5) |
*Intervaly spolehlivosti byly vypočteny bez úpravy podle pohlaví a oblasti, které byly zařazeny v primární analýze za použití logistických modelů.
Přínos kombinované terapie s aprepitantem u celé hodnocené populace byl dán zejména výsledky pozorovanými u pacientů se slabou kontrolou při standardním režimu, jako jsou ženy, i když výsledky byly numericky lepší bez ohledu na věk, typ tumoru nebo pohlaví. Úplné odpovědi bylo dosaženo u 209/324 (65 %) v případě režimu s aprepitantem a u 161/320 (50 %) při standardní terapii u žen a u mužů šlo v případě režimu s aprepitantem u 83/101 (82 %) a při standardní terapii u 68/87 (78 %).
Pediatrická populace
V randomizované, dvojitě zaslepené klinické studii, kontrolované aktivním komparátorem, která zahrnovala 302 dětí a dospívajících (ve věku 6 měsíců až 17 let), kterým se podávala středně silně nebo vysoce emetogenní chemoterapie, se režim s aprepitantem porovnával s kontrolním režimem prevence CINV. Účinnost režimu s aprepitantem byla hodnocena při jednom cyklu (cyklus 1). Pacienti měli v následných cyklech (volitelné cykly 2 až 6) možnost dostávat aprepitant v otevřeném uspořádání; v těchto volitelných cyklech však účinnost hodnocena nebyla. Režim s aprepitantem u dospívajících ve věku 12 až 17 let (N = 47) sestával z přípravku EMEND tobolky 125 mg podávaného perorálně 1. den a z 80 mg/den 2. a 3. den v kombinaci s ondansetronem 1. den. Režim s aprepitantem u dětí ve věku 6 měsíců až méně než 12 let (N = 105) sestával z přípravku EMEND prášek pro
perorální suspenzi podávaného v dávce 3,0 mg/kg (do dávky 125 mg) perorálně 1. den a v dávce
2,0 mg/kg (do dávky 80 mg) perorálně 2. a 3. den v kombinaci s ondansetronem 1. den. Kontrolní režim u dospívajících ve věku 12 až 17 let (N = 48) a dětí ve věku 6 měsíců až méně než 2 roky (N = 102) sestával z placeba místo aprepitantu 1., 2. a 3. den v kombinaci s ondansetronem 1. den. Přípravek EMEND nebo placebo a ondansetron se podávaly 1 hodinu, respektive 30 minut před zahájením chemoterapie. Jako součást antiemetického režimu pro pediatrické pacienty byl u obou věkových skupin povolen intravenózní dexamethason, a to podle rozhodnutí lékaře. U pediatrických pacientů léčených aprepitantem bylo požadováno snížení (50%) dávky dexamethasonu. U pediatrických pacientů léčených kontrolním režimem nebylo snížení dávky požadováno. Z pediatrických pacientů používalo v 1. cyklu ve skupině léčené režimem s aprepitantem dexamethason 29 % zúčastněných a ve skupině s kontrolním režimem 28 % zúčastněných.
Antiemetická aktivita přípravku EMEND byla hodnocena během 5denního (120 hodin) období po zahájení chemoterapie 1. den. Primárními kritérii hodnocení byla úplná odpověď v pozdní fázi (25 až 120 hodin po zahájení chemoterapie) v 1. cyklu. Souhrn klíčových výsledků studie je uveden v Tabulce 4.
Tabulka 4
Počet (%) pediatrických pacientů s úplnou odpovědí a bez zvracení podle léčebné skupiny a fáze - 1.
cyklus (populace všech zařazených pacientů)
|
Režim s aprepitanten n/m (%) |
Kontrolní režim n/m (%) | |
|
PRIMÁRNÍ KRITÉRIUM HODNOCENÍ | ||
|
Úplná odpověď * - pozdní fáze |
77/152 (50,7)t |
39/150 (26,0) |
|
DALŠÍ PŘEDEM DEFINOVANÁ KRITÉRIA HODNOCENÍ | ||
|
Úplná odpověď * - akutní fáze |
101/152 (66,4); |
78/150 (52,0) |
|
Úplná odpověď - úplná fáze |
61/152 (40,1)t |
30/150 (20,0) |
|
i* Bez zvracení § - úplná fáze |
71/152 (46,7)t |
32/150 (21,3) |
|
*Úplná odpověď = žádné zvracení ani říhání nebo zvedání žaludku naprázc záchranné medikace. Tp < 0,01 v porovnání s kontrolním režimem *p < 0,05 v porovnání s kontrolním režimem §Žádné zvracení = žádné zvracení ani říhání nebo zvedání žaludku naprázdno n/m = počet pacientů s požadovanou odpovědí/počet pacientů zařazených v daný o Akutní fáze: 0 až 24 hodin po zahájení chemoterapie. Pozdní fáze: 25 až 120 hodin po zahájení chemoterapie. Úplná fáze: 0 až 120 hodin po zahájení chemoterapie. |
no a žádné použití kamžik. | |
Odhadovaná doba do prvního zvracení po zahájení léčby chemoterapií byla u režimů s aprepitantem delší (odhadovaný medián času do prvního zvracení byl 94,5 hodiny) v porovnání se skupinou s kontrolním režimem (odhadovaný medián času do prvního zvracení byl 26,0 hodiny), jak je uvedeno Kaplan-Meierovými křivkami na Grafu 2.
Graf 2
Dob a do první epizody zvracení od začátku podávání chemoterapie pediatrickým pacientům v celkové
fázi 1. cyklu (populace všech zařazených pacientů)
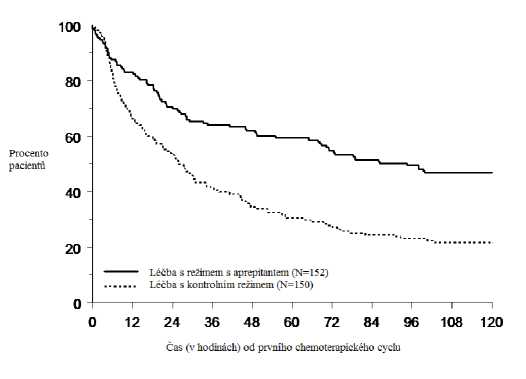
Analýza účinnosti u subpopulací v 1. cyklu prokázala, že režim s aprepitantem poskytoval lepší kontrolu než kontrolní režim, pokud jde o kritérium hodnocení úplná odpověď, a to bez ohledu na věkovou kategorii, pohlaví, podání dexamethasonu k antiemetické profylaxi a emetogenitu chemoterapie.
5.2 Farmakokinetické vlastnosti
Farmakokinetika aprepitantu má nelineární charakter. Clearance i absolutní biologická dostupnost se se zvyšováním dávky snižují.
Absorpce
Střední hodnota biologické dostupnosti perorálně podaného aprepitantu činí 67 % u 80mg tobolky a 59 % u 125mg tobolky. Průměrné maximální plazmatické koncentrace (Cmax) aprepitantu byly dosaženy přibližně po 4 hodinách (tmax). Perorální podání tobolky spolu se standardní snídaní s kalorickou hodnotou přibližně 800 kcal vedlo až k 40% zvýšení hodnoty AUC aprepitantu. Toto zvýšení se nepovažuje za klinicky významné.
Farmakokinetika aprepitantu má nelineární charakter v celém rozsahu klinických dávek. U mladých zdravých dospělých jedinců bylo zvýšení hodnoty AUC0-<X) o 26 % větší, než byla proporcionální dávka mezi jednotlivými 80mg a 125mg dávkami podanými v nasyceném stavu.
Po perorálním podání jedné 125mg dávky přípravku EMEND 1. den a dávky 80 mg jednou denně
2. a 3. den dosáhla hodnota AUC0-24hod. (střední hodnota ± standardní odchylka) 19,6 ± 2,5 pg • hod/ml 1. den a 21,2 ± 6,3 pg • hod/ml 3. den. Hodnota Cmax činila 1. den 1,6 ± 0,36 pg a 3. den 1,4 ± 0,22 pg.
Distribuce
Aprepitant se ve vysoké míře váže na proteiny, se střední hodnotou 97 %. Geometrická střední hodnota zdánlivého distribučního objemu v rovnovážném stavu (Vdss) je u člověka přibližně 66 litrů.
Biotransformace
Aprepitant prochází rozsáhlou biotransformací. U zdravých mladých dospělých jedinců vykazuje aprepitant po dobu 72 hodin po jednorázové intravenózní aplikaci 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, v plazmě přibližně 19 % radioaktivity, což ukazuje na značnou přítomnost metabolitů v plazmě. V lidské plazmě bylo zjištěno dvanáct metabolitů aprepitantu. Metabolismus aprepitantu probíhá ve velké míře cestou oxidace v morfolinovém kruhu a jeho postranních řetězcích a výsledné metabolity jsou pouze slabě aktivní. In vitro studie s lidskými jaterními mikrozómy ukázaly, že aprepitant se biotransformuje primárně cestou CYP3A4, případně s malým podílem CYP1A2 a CYP2C19.
Aprepitant se nevylučuje močí v nezměněné podobě. Metabolity se vylučují močí a žlučí ve stolici.
Po jednorázově intravenózně aplikované dávce 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, zdravým jedincům bylo 57 % radioaktivity zjištěno v moči a 45 % ve stolici.
Plazmatická clearance aprepitantu závisí na dávce, se zvyšující se dávkou se snižuje a v rozmezí terapeutických dávek se pohybovala přibližně na hodnotách od 60 do 72 ml/min. Terminální poločas se pohyboval v rozmezí přibližně od 9 do 13 hodin.
Farmakokinetika u speciálních populací
Starší osoby: Po perorálním podání jedné 125mg dávky aprepitantu 1. den a dávky 80 mg jednou denně 2. až 5. den byla hodnota AUC aprepitantu 1. den o 21 % vyšší a 5. den o 36 % vyšší u starších jedinců (> 65 let) ve srovnání s mladšími dospělými. Hodnota Cmax byla u starších ve srovnání s mladšími dospělými 1. den o 10 % vyšší a 5. den o 24 % vyšší. Tyto rozdíly se nepovažovaly za klinicky významné. U starších pacientů není třeba dávku nijak upravovat.
Pohlaví: Po perorálním podání jediné 125mg dávky aprepitantu byla hodnota Cmax aprepitantu u žen ve srovnání s muži o 16 % vyšší. Poločas aprepitantu je u žen ve srovnání s muži o 25 % nižší a k dosažení tnax dochází zhruba ve stejnou dobu. Tyto rozdíly nejsou považovány za klinicky významné. Dávku přípravku EMEND není nutno podle pohlaví pacienta nijak upravovat.
Porucha funkce jater: Mírná porucha funkce jater (Childovo-Pughovo hodnocení, třída A) farmakokinetiku aprepitantu v klinicky významné míře neovlivňuje. U pacientů s mírnou poruchou funkce jater není třeba dávkování nijak upravovat. Z dostupných dat nelze činit žádné závěry ohledně vlivu středně těžké poruchy funkce jater (Childovo-Pughovo hodnocení, třída B) na farmakokinetiku aprepitantu. K dispozici nejsou žádné klinické ani farmakokinetické údaje od pacientů s těžkou poruchou funkce jater (Childovo-Pughovo hodnocení, třída C).
Porucha funkce ledvin: Pacientům s těžkou poruchou funkce ledvin (CrCl < 30 ml/min) a pacientům s terminálním renálním onemocněním (end stage renal disease, ESRD) s potřebou hemodialýzy byla podána jednorázová 240mg dávka aprepitantu.
U pacientů s těžkou poruchou funkce ledvin se hodnota AUC0_W celkového aprepitantu (nevázaného i vázaného na proteiny) ve srovnání se zdravými jedinci snížila o 21 % a hodnota Cmax se snížila o 32 %. U pacientů s ESRD podstupujících hemodialýzu se hodnota AUC0_<» celkového aprepitantu snížila o 42 % a hodnota Cmax se snížila o 32 %. Vzhledem k mírnému poklesu vazby aprepitantu na proteiny u pacientů s renálním onemocněním nebyla hodnota AUC farmakologicky aktivní části nenavázaného léku u pacientů s poruchou funkce ledvin ve srovnání se zdravými jedinci významně ovlivněna. Hemodialýza prováděná 4 nebo 48 hodin po podání dávky neměla na farmakokinetiku aprepitantu významný účinek; v dialyzátu bylo zjištěno méně než 0,2 % dávky.
U pacientů s poruchou funkce ledvin ani u pacientů s ESRD podstupujících hemodialýzu není zapotřebí dávkování přípravku EMEND nijak upravovat.
Pediatrická populace: v rámci 3denního režimu se u většiny pacientů dávkováním aprepitantu v tobolkách (125/80/80 mg) u dospívajících pacientů (ve věku 12 až 17 let) první den dosáhlo AUC0-24hod nad 17 ^g^hod/ml s koncentracemi (Cmin) na konci 2. a 3. dne nad 0,4 ^g/ml. Medián maximálních plazmatických koncentrací (Cmax) byl 1. den přibližně 1,3 ^g/ml a tato koncentrace se objevila za přibližně 4 hodiny. V rámci 3denního režimu se u většiny pacientů dávkováním aprepitantu v prášku pro přípravku perorální suspenze (3/2/2 mg/kg) u pacientů ve věku 6 měsíců až méně než 12 let první den dosáhlo AUC0.24hod nad 17 ^g^hod/ml s koncentracemi (Cmin) na konci 2. a
3. dne nad 0,1 ^g/ml. Medián maximálních plazmatických koncentrací (Cmax) byl první den přibližně
1,2 (ig/ml a tato koncentrace se objevila za 5 až 7 hodin.
Populační farmakokinetická analýza aprepitantu u pediatrických pacientů (ve věku 6 měsíců až 17 let) naznačuje, že pohlaví a rasa na farmakokinetiku aprepitantu klinicky významný vliv nemají.
Vztah mezi koncentrací a účinkem
Studie využívající pozitronovou emisní tomografii (PET) s použitím vysoce specifického značení receptorů NK u zdravých mladých mužů ukázaly, že aprepitant prostupuje do mozku a obsazuje receptory NK v závislosti na dávce a koncentraci v plazmě. Předpokládá se, že plazmatické koncentrace aprepitantu dosažené při 3-denním léčebném režimu s přípravkem EMEND u dospělých zajišťují více než 95% obsazení receptorů NK v mozku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické údaje získané na základě konvenčních studií toxicity, genotoxicity, hodnocení kancerogenního potenciálu, reprodukční a vývojové toxicity po jednorázovém a opakovaném podání přípravku neodhalily žádné zvláštní riziko pro člověka. Je však nutno mít na paměti, že systémová expozice u hlodavců byla podobná nebo dokonce nižší, než byla terapeutická expozice u člověka při dávce 125 mg/80 mg. I když nebyly v reprodukčních studiích s expozičními hodnotami pro člověka zjištěny žádné nežádoucí účinky, nelze zejména na základě hodnot expozice použitých u zvířat dostatečně posoudit riziko pro člověka.
Ve studii juvenilní toxicity na potkanech, kterým se podával aprepitant od 10. do 63. postnatálního dne, docházelo u samic k časnému vaginálnímu otevírání od dávek 250 mg/kg dvakrát denně a u samců k opožděné prepuciální separaci, a to od dávek 10 mg/kg dvakrát denně. Nebyl zde žádný odstup od klinicky relevantní expozice. Nedošlo k žádným účinkům souvisejícím s léčbou ohledně páření, fertility či embryonálního/fetálního vývoje a nevyskytly se žádné patologické změny reprodukčních orgánů. Ve studii juvenilní toxicity na psech ošetřovaných od 14. do 42. postnatálního dne byla u samců při dávce 6 mg/kg/den pozorována snížená hmotnost varlat a velikost Leydigových buněk a u samic při dávkách od 4 mg/kg/den byla pozorována zvýšená hmotnost dělohy, hypertrofie dělohy a krčku a otok vaginálních tkání. Nebyl zde žádný odstup od klinicky relevantní expozice aprepitantu. Při krátkodobé léčbě podle doporučeného dávkovacího režimu se klinická relevance těchto zjištění nepovažuje za pravděpodobnou.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Obsah tobolky Sacharóza
Mikrokrystalická celulóza (E 460) Hyprolóza (E 463) Natrium-lauryl-sulfát
Obal tobolky (125 mg)
Želatina
Oxid titaničitý (E 171)
Červený oxid železitý (E 172)
Žlutý oxid železitý (E 172)
Obal tobolky (80 mg)
Želatina
Oxid titaničitý (E 171)
Inkoust na potisk Šelak
Hydroxid draselný Černý oxid železitý (E 172)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
4 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
6.5 Druh obalu a obsah balení
Na trhu jsou dostupné různé velikosti balení obsahující rozdílné síly.
Hliníkový blistr obsahující jednu 80mg tobolku.
Hliníkový blistr obsahující dvě 80mg tobolky.
5 hliníkových blistrů, každý obsahující jednu 80mg tobolku.
Hliníkový blistr obsahující jednu 125mg tobolku.
5 hliníkových blistrů, každý obsahující jednu 125mg tobolku.
Hliníkový blistr obsahující jednu 125mg tobolku a dvě 80mg tobolky.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO
EU/1/03/262/001
EU/1/03/262/002
EU/1/03/262/003
EU/1/03/262/004
EU/1/03/262/005
EU/1/03/262/006
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. listopadu 2003 Datum prodloužení registrace: 22. září 2008
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
EMEND 125 mg prášek pro perorální suspenzi
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jeden sáček obsahuje aprepitantum 125 mg. Po rekonstituci obsahuje 1 ml perorální suspenze aprepitantum 25 mg.
Pomocné látky se známým účinkem
Jeden sáček obsahuje přibližně 125 mg sacharózy a 468,7 mg laktózy (bezvodé).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro perorální suspenzi. Růžový až světle růžový prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Prevence nauzey a zvracení spojených s vysoce a středně silně emetogenní protinádorovou chemoterapií u dětí, batolat a kojenců ve věku 6 měsíců až méně než 12 let.
EMEND prášek pro perorální suspenzi se podává jako součást kombinované terapie (viz bod 4.2).
4.2 Dávkování a způsob podání
Dávkování
Pediatrická _ populace
Kojenci, batolata a děti (ve věku 6 měsíců až méně než 12 let s hmotností od 6 kg)
Přípravek EMEND se podává po dobu 3 dní jako součást léčebného režimu, který zahrnuje antagonistu 5-HT3. Doporučená dávka přípravku EMEND prášek pro perorální suspenzi je založena na hmotnosti, jak je specifikováno v tabulce níže. Přípravek EMEND se podává perorálně 1 hodinu před chemoterapií 1., 2. a 3. den. Pokud se 2. a 3. den nepodává žádná chemoterapie, má být přípravek EMEND podáván ráno. Ohledně správného dávkování zvoleného antagonisty 5-HT3 nahlédněte do jeho souhrnu údajů o přípravku (SPC). Pokud se s přípravkem EMEND podává kortikosteroid, jako je dexamethason, musí se podat 50 % obvyklé dávky kortikosteroidu (viz body 4.5 a 5.1).
Doporučená dávka a objem přípravku EMEND perorální suspenze u pediatrických pacientů
ve věku 6 měsíců až méně než 12 let
|
Tělesná hmotnost |
Dávkový objem perorálně podávané sus |
penze | |
|
1. den |
2. den |
3. den | |
|
méně než 6 kg |
Nedoporučuje se | ||
|
6 kg až méně než 8 kg |
1 ml (25 mg) |
0,6 ml (15 mg) |
0,6 ml (15 mg) |
|
8 kg až méně než 10 kg |
1,2 ml (30 mg) |
0,8 ml (20 mg) |
0,8 ml (20 mg) |
|
10 kg až méně než 12 kg |
1,4 ml (35 mg) |
1 ml (25 mg) |
1 ml (25 mg) |
|
12 kg až méně než 15 kg |
1,8 ml (45 mg) |
1,2 ml (30 mg) |
1,2 ml (30 mg) |
|
15 kg až méně než 20 kg |
2,4 ml (60 mg) |
1,6 ml (40 mg) |
1,6 ml (40 mg) |
|
20 kg až méně než 25 kg |
3 ml (75 mg) |
2 ml (50 mg) |
2 ml (50 mg) |
|
25 kg až méně než 30 kg |
3,6 ml (90 mg) |
2,4 ml (60 mg) |
2,4 ml (60 mg) |
|
30 kg a více |
Obsah (125 mg) mísicí nádobky natáhněte do perorálního dávkovače (přibližně 5 ml) |
3,2 ml (80 mg) |
3,2 ml (80 mg) |
Účinnost 125mg prášku pro perorální suspenzi nebyla u dospívajících ve věku 12 let a starších stanovena. Pro dospívající ve věku 12 až 17 let je k dispozici přípravek EMEND ve formě tobolek obsahujících 80 nebo 125 mg aprepitantu.
Bezpečnost a účinnost přípravku EMEND prášek pro perorální suspenzi u kojenců mladších 6 měsíců nebo s hmotností menší než 6 kg nebyla stanovena. K dispozici nejsou žádné údaje.
Obecně
Údaje o účinnosti při kombinaci s kortikosteroidy a antagonisty 5-HT3 jsou omezené. Další informace o současném podávání s kortikosteroidy jsou uvedeny v bodu 4.5. U současně podávaných léčivých přípravků, antagonistů 5-HT3 nahlédněte do jejich SPC.
Zvláštní _ populace Pohlaví
S ohledem na pohlaví není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce ledvin
U pacientů s poruchou funkce ledvin ani u pacientů v terminálním stádiu renálního onemocnění podstupujících hemodialýzu není nutno dávku nijak upravovat (viz bod 5.2).
Porucha funkce jater
U pacientů s mírnou poruchou funkce jater není nutno dávku nijak upravovat. Pokud se týče pacientů se středně těžkou poruchou funkce jater, je k dispozici pouze omezené množství dat a nejsou dostupné žádné údaje od pacientů s těžkou poruchou funkce jater. Těmto pacientům se aprepitant musí podávat opatrně (viz body 4.4 a 5.2).
Způsob podání
Perorální suspenzi je možno užívat bez ohledu na jídlo..
Podrobnosti o přípravě a podávání suspenze, viz bod 6.6.
4.3 Kontraindikace
Hypersenzitivita na léčivou látku nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
Současné podávání s pimozidem, terfenadinem, astemizolem nebo cisapridem (viz bod 4.5).
4.4 Zvláštní upozornění a opatření pro použití Pacienti se středně těžkou až těžkou poruchou funkce jater
Pro pacienty se středně těžkou poruchou funkce jater existuje pouze omezené množství dat a žádné údaje nejsou k dispozici pro pacienty s těžkou poruchou funkce jater. U těchto pacientů je nutno přípravek EMEND používat s opatrností (viz bod 5.2).
Interakce na CYP3A4
EMEND je nutno podávat s opatrností pacientům současně užívajícím perorálně podávané léčivé látky, které se primárně metabolizují cestou CYP3A4 a které mají úzké terapeutické rozmezí, jako je cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, deriváty námelových alkaloidů, fentanyl a chinidin (viz bod 4.5). Navíc je nutno k podávání současně s irinotekanem přistupovat s obzvláštní opatrností, protože uvedená kombinace by mohla mít za následek zvýšenou toxicitu.
Současné podávání s warfarinem (substrát CYP2C9)
U pacientů dlouhodobě léčených warfarinem je nutno hodnotu mezinárodního normalizovaného poměru (INR) pozorně sledovat během léčby přípravkem EMEND a po dobu 14 dnů po každé 3-denní léčbě přípravkem EMEND (viz bod 4.5).
Současné podávání s hormonální antikoncepcí
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a ještě 2 měsíce po poslední dávce přípravku EMEND je třeba používat alternativní nehormonální záložní antikoncepční metody (viz bod 4.5).
Pomocné látky
Přípravek EMEND prášek pro perorální suspenzi obsahuje sacharózu a laktózu. Pacienti se vzácnými dědičnými problémy s intolerancí fruktózy nebo galaktózy, malabsorpcí glukózy-galaktózy, vrozeným deficitem laktázy nebo deficitem sacharáza-izomaltázy nemají tento přípravek užívat.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Aprepitant (125 mg/80 mg) je substrát, středně silný inhibitor a induktor CYP3A4. Aprepitant je také induktor CYP2C9. Během léčby přípravkem EMEND je inhibován CYP3A4. Po ukončení léčby přípravkem EMEND dochází k přechodné mírné indukci CYP2C9, CYP3A4 a glukuronidace. Nezdá se, že by aprepitant interagoval s P glykoproteinovým transportérem, jak naznačuje nepřítomnost interakce aprepitantu s digoxinem.
Účinek aprepitantu na farmakokinetiku dalších léčivých látek Inhibice CYP3A4
Jako středně silný inhibitor CYP3A4 může aprepitant (125 mg/80 mg) zvýšit plazmatické koncentrace současně podávaných léčivých látek, které se metabolizují cestou CYP3A4. Celková expozice perorálně podávaným substrátům CYP3A4 se může zvýšit přibližně 3krát během 3denní léčby přípravkem EMEND; předpokládá se, že účinek aprepitantu na plazmatické koncentrace intravenózně podávaných substrátů CYP3A4 bude menší. Přípravek EMEND se nesmí podávat současně s pimozidem, terfenadinem, astemizolem nebo cisapridem (viz bod 4.3). Inhibice CYP3A4 aprepitantem by mohla mít za následek zvýšené plazmatické koncentrace uvedených léčivých látek, což může vyvolat závažné nebo život ohrožující reakce. Při současném podávání přípravku EMEND a perorálně podávaných léčivých látek, které jsou metabolizovány převážně prostřednictvím CYP3A4 a kte ré mají úzké terapeutické rozmezí, jako je cyklosporin, takrolimus, sirolimus, everolimus, alfentanil, diergotamin, ergotamin, fentanyl a chinidin, se doporučuje opatrnost (viz bod 4.4).
Kortikosteroidy
Dexamethason: Obvyklou dávku perorálně podaného dexamethasonu je nutno při současném podávání s přípravkem EMEND v režimu 125 mg/80 mg snížit přibližně o 50 %. Dávka dexamethasonu v klinických studiích nauzey a zvracení vyvolaných chemoterapií byla vybrána s ohledem na interakce léčivých látek (viz bod 4.2). Pokud se EMEND podával v léčebném režimu v dávce 125 mg spolu s dexamethasonem 20 mg perorálně první den, a poté v dávce 80 mg/den spolu s dexamethasonem 8 mg perorálně druhý až pátý den, došlo první a pátý den k 2,2násobnému zvýšení hodnoty AUC dexamethasonu, který je substrátem CYP3A4.
Methylprednisolon: Při současném podávání s přípravkem EMEND v režimu 125 mg/80 mg je třeba obvyklou dávku intravenózně aplikovaného methylprednisolonu snížit přibližně o 25 %, a obvyklou dávku perorálně podávaného methylprednisolonu přibližně o 50 %. Pokud se EMEND podával v léčebném režimu v dávce 125 mg první den a 80 mg/den 2. a 3. den, zvýšil hodnotu AUC methylprednisolonu, který je substrátem CYP3A4, 1. den 1,3krát a 3. den 2,5krát v případě, že se methylprednisolon podával současně intravenózně v dávce 125 mg 1. den a perorálně v dávce 40 mg 2. a 3. den.
V průběhu kontinuální léčby methylprednisolonem se může hodnota AUC methylprednisolonu koncem 2týdenního období po zahájení podávání přípravku EMEND snížit v důsledku indukčního účinku aprepitantu na CYP3A4. Lze očekávat, že tento účinek bude u perorálně podávaného methylprednisolonu výraznější.
Chemoterapeutické léčivé přípravky
Ve farmakokinetických studiích neměl EMEND při podávání v režimu 125 mg 1. den a 80 mg/den 2. a 3. den vliv na farmakokinetiku docetaxelu podaného intravenózně 1. den nebo vinorelbinu podaného intravenózně 1. nebo 8. den. Protože účinek přípravku EMEND na farmakokinetiku perorálně podávaných substrátů CYP3A4 je větší než účinek přípravku EMEND na farmakokinetiku intravenózně podávaných substrátů CYP3A4, nelze vyloučit interakce s perorálně podávanými chemoterapeutickými léčivými přípravky, které se biotransformují převážně nebo částečně cestou CYP3A4 (např. etoposid, vinorelbin). U pacientů dostávajících léčivé přípravky, které se metabolizují především nebo částečně CYP3A4, se doporučuje opatrnost, přičemž může být vhodné tyto pacienty dodatečně sledovat (viz bod 4.4). Po uvedení na trh byly hlášeny při současném podávání aprepitantu a ifosfamidu případy neurotoxicity, což je potenciální nežádoucí účinek ifosfamidu.
Imunosupresiva
V průběhu 3denního režimu léčby nauzey a zvracení vyvolaných chemoterapií se předpokládá přechodné střední zvýšení následované mírným poklesem expozice imunosupresivům metabolizovaným prostřednictvím CYP3A4 (např. cyklosporin, takrolimus, everolimus a sirolimus).
S ohledem na krátké trvání 3denního režimu a na časově závislé omezené změny expozice se během 3 dnů současného podávání s přípravkem EMEND snížení dávky imunosupresiv nedoporučuje.
Midazolam
Při současném podávání midazolamu nebo jiných benzodiazepinů metabolizovaných cestou CYP3A4 (alprazolam, triazolam) spolu s přípravkem EMEND (125 mg/80 mg) je nutno vzít v úvahu možné účinky zvýšených plazmatických koncentrací těchto léčivých přípravků.
EMEND zvýšil hodnotu AUC midazolamu, který je citlivým substrátem CYP3A4, 2,3krát 1. den a 3,3krát 5. den, pokud byl midazolam podán perorálně v jediné 2mg dávce 1. a 5. den léčebného režimu s přípravkem EMEND v dávce 125 mg 1. den a 80 mg/den 2. až 5. den.
V jiné studii s intravenózní aplikací midazolamu se EMEND podával v dávce 125 mg 1. den
a 80 mg/den 2. a 3. den a midazolam v dávce 2 mg se aplikoval intravenózně před zahájením 3denního léčebného režimu s přípravkem EMEND a 4., 8., a 15. den. EMEND zvýšil hodnotu AUC midazolamu o 25 % 4. den a snížil hodnotu AUC midazolamu o 19 % 8. den a o 4 % 15. den. Tyto účinky nebyly považovány za klinicky významné.
Ve třetí studii s intravenózní a perorální aplikací midazolamu se EMEND podával v dávce 125 mg 1. den a 80 mg/den 2. a 3. den společně s ondansetronem 32 mg 1. den, dexamethasonem 12 mg 1. den a 8 mg 2. až 4. den. Tato kombinace (tedy EMEND, ondansetron a dexamethason) snížila hodnotu AUC perorálního midazolamu o 16 % 6. den, o 9 % 8. den, o 7 % 15. den a o 17 % 22. den. Tyto účinky nebyly považovány za klinicky významné.
Byla provedena dodatečná studie intravenózního podávání midazolamu a přípravku EMEND. Midazolam v dávce 2 mg byl podán intravenózně 1 hodinu po perorálně podané jedné 125mg dávce přípravku EMEND. Plazmatická hodnota AUC midazolamu se zvýšila 1,5krát. Tento účinek nebyl považován za klinicky významný.
Indukce
Jako mírný induktor CYP2C9, CYP3A4 a glukuronidace může aprepitant během dvou týdnů po zahájení a léčbě snižovat plazmatické koncentrace substrátů eliminovaných těmito cestami. Tento účinek se může projevit až po skončení třídenní léčby přípravkem EMEND. U substrátů CYP2C9 a CYP3A4 je indukce přechodná s dosažením maxima účinku 3 až 5 dní po skončení třídenní léčebné kúry přípravkem EMEND. Účinek se zachovává po dobu několika dní, poté se zvolna ztrácí a dva týdny po skončení léčby přípravkem EMEND je již klinicky nevýznamný. Mírnou indukci glukuronidace lze pozorovat i při perorálním podávání aprepitantu v dávce 80 mg po dobu 7 dní.
Údaje ohledně účinku na CYP2C8 a CYP2C19 nejsou k dispozici. Opatrnost se doporučuje v případech, kdy se v daném období podávají warfarin, acenokumarol, tolbutamid, fenytoin nebo jiné léčivé látky, o nichž je známo, že jsou metabolizovány CYP2C9.
Warfarin
U pacientů dlouhodobě léčených warfarinem je třeba pozorně sledovat protrombinový čas (INR) během léčby přípravkem EMEND a po dobu 2 týdnů po každé 3denní léčbě přípravkem EMEND z důvodu chemoterapií vyvolané nauzey a zvracení (viz bod 4.4). Jestliže se podala jediná 125mg dávka přípravku EMEND 1. den a dávka 80 mg/den 2. a 3. den zdravým jedincům ve stabilizovaném stavu při dlouhodobé léčbě warfarinem, neměl EMEND 3. den žádný účinek na hodnotu plazmatické AUC pro R(+) ani S(-) warfarin, nicméně 5 dní po skončení podávání přípravku EMEND, ale došlo ke 34% snížení dolní hodnoty koncentrace S(-) warfarinu (substrátu CYP2C9) spolu se 14% poklesem hodnoty INR.
Tolbutamid
Pokud se EMEND podával v dávce 125 mg 1. den a v dávce 80 mg/den 2. a 3. den, snížil 4. den hodnotu AUC tolbutamidu (substrátu CYP2C9) o 23 %, 8. den o 28 %, a 15. den o 15 % v případě, že se jediná 500mg dávka tolbutamidu podala perorálně před zahájením 3denní kúry s přípravkem EMEND a pak 4., 8., a 15. den.
Hormonální antikoncepce
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a po dobu 28 dní poté snížena. Během léčby přípravkem EMEND a ještě 2 měsíce po poslední dávce přípravku EMEND je třeba používat alternativní nehormonální záložní antikoncepční metody.
V klinické studii byly 1. až 21. den podávány jednorázové dávky perorální antikoncepce obsahující ethinylestradiol a norethisteron spolu s přípravkem EMEND, podávaným v režimu v dávce 125 mg 8. den a 80 mg/den 9. a 10. den s ondansetronem v dávce 32 mg podaným intravenózně 8. den a dexamethasonem v dávce 12 mg podaným perorálně 8. den a dále 8 mg/den 9., 10. a 11. den. V této studii se minimální koncentrace ethinylestradiolu snížily 9. až 21. den až o 64 % a minimální koncentrace norethisteronu až o 60 %.
Antagonisté 5-HT3
V klinických studiích lékových interakcí nevykazoval aprepitant žádné klinicky významné účinky na farmakokinetiku ondansetronu, granisetronu nebo hydrodolasetronu (aktivní metabolit dolasetronu).
Účinky jiných léčivých přípravků na farmakokinetiku aprepitantu
K současnému podávání přípravku EMEND s léčivými látkami, které inhibují aktivitu CYP3A4 (např. ketokonazol, itrakonazol, vorikonazol, posakonazol, klarithromycin, telithromycin, nefazodon a inhibitory proteázy), je třeba přistupovat opatrně, protože se předpokládá, že tato kombinace povede k několikanásobnému zvýšení plazmatické koncentrace aprepitantu (viz bod 4.4).
Je nutno se vyvarovat současného podávání přípravku EMEND s léčivými látkami, které silně indukují aktivitu CYP3A4 (např. s rifampicinem, fenytoinem, karbamazepinem, fenobarbitalem), protože tato kombinace vede ke snížení plazmatických koncentrací aprepitantu, což může vést ke snížení účinnosti přípravku EMEND. Současné podávání přípravku EMEND s bylinnými přípravky obsahujícími třezalku tečkovanou (Hypericumperforatum) se nedoporučuje.
Ketokonazol
Jestliže se podal aprepitant v jediné 125mg dávce 5. den 10denního léčebného režimu se silným inhibitorem CYP3A4 ketokonazolem podaným v dávce 400 mg/den, zvýšila se hodnota AUC aprepitantu přibližně 5krát a střední hodnota terminálního poločasu aprepitantu se zvýšila přibližně 3krát.
Rifampicin
Jestliže se podal aprepitant v jediné 375 mg dávce 9. den 14denního léčebného režimu se silným induktorem CYP3A4 rifampicinem podaným v dávce 600 mg/den, snížila se hodnota AUC aprepitantu o 91 % a střední hodnota terminálního poločasu aprepitantu se snížila o 68 %.
Pediatrická populace
Studie interakcí byly provedeny pouze u dospělých.
4.6 Fertilita, těhotenství a kojení
Antikoncepce u mužů a žen
Účinnost hormonální antikoncepce může být během podávání přípravku EMEND a 28 dní poté snížena. Během léčby přípravkem EMEND a 2 měsíce po poslední dávce přípravku EMEND je nutno používat alternativní nehormonální záložní antikoncepční metody (viz body 4.4 a 4.5).
Nejsou dostupné žádné klinické údaje týkající se těhotenství vystavených aprepitantu. Potenciální reprodukční toxicita aprepitantu nebyla zcela stanovena, protože míry expozice vyšší než hodnoty terapeutické expozice u člověka při dávce 125 mg/80 mg nebylo možno ve studiích se zvířaty dosáhnout. Tyto studie nenaznačily žádné přímé ani nepřímé škodlivé účinky na průběh těhotenství, embryonální/fetální vývoj, porod nebo postnatální vývoj (viz bod 5.3). Možné účinky na reprodukci změn v regulaci neurokininů nejsou známy. EMEND nemá být během těhotenství podáván, pokud to není nezbytně nutné.
Kojení
Aprepitant se vylučuje do mléka kojících potkanů. Není známo, zda se aprepitant vylučuje do lidského mateřského mléka, a proto se kojení během léčby přípravkem EMEND nedoporučuje.
Fertilita
Potenciál aprepitantu navodit účinky na fertilitu nebyl dosud plně popsán, protože expozičních hladin přesahujících terapeutickou expozici u lidí nemohlo být ve studiích na zvířatech dosaženo. Tyto studie fertility neukazují na přímé nebo nepřímé škodlivé účinky ohledně páření, fertility, embryonálního/fetálního vývoje či počtu a motility spermií (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek EMEND může mít mírný vliv na schopnost jezdit na kole nebo obsluhovat stroje. Po užití přípravku EMEND se mohou objevit závratě a únava (viz bod 4.8).
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Profil bezpečnosti aprepitantu byl hodnocen přibližně u 6 500 dospělých ve více než 50 studiích a 184 dětí a dospívajících ve 2 kontrolovaných pediatrických klinických studiích.
Nejčastějšími nežádoucími účinky, uváděnými ve vyšším výskytu u dospělých léčených režimem s aprepitantem než standardní terapií, u pacientů léčených vysoce emetogenní chemoterapií byly: škytavka (4,6 % oproti 2,9 %), zvýšení ALT (2,8 %, oproti 1,1 %), dyspepsie (2,6 %, oproti 2,0 %), zácpa (2,4 %, oproti 2,0 %), bolest hlavy (2,0 %, oproti 1,8 %) a snížení chuti k jídlu (2,0 %, oproti 0,5 %). Nejčastějším nežádoucím účinkem uváděným ve vyšším výskytu u pacientů léčených režimem s aprepitantem než standardní terapií u dospělých léčených středně emetogenní chemoterapií byla únava (1,4 %, oproti 0,9 %).
Nejčastějšími nežádoucími účinky hlášenými s vyšší incidencí u pediatrických pacientů léčených tímto režimem aprepitantu než u kontrolního režimu během léčby emetogenní protinádorovou chemoterapií byla škytavka (3,3 % versus 0,0 %) a zarudnutí (1,1 % versus 0,0 %).
Tabulkový seznam nežádoucích účinků
Následující nežádoucí účinky byly pozorovány v souhrnné analýze studií s vysoce emetogenní chemoterapií a středně emetogenní chemoterapií u pacientů léčených režimem s aprepitantem s vyšší incidencí než při standardní terapii nebo po uvedení přípravku na trh. Kategorie frekvence uvedené v tabulce jsou založeny na studiích u dospělých; pozorované frekvence v pediatrických studiích byly podobné nebo nižší, není-li v tabulce uvedeno. Některé méně časté nežádoucí účinky v dospělé populaci nebyly pozorovány v pediatrických studiích
Frekvence jsou definovány jako: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1 000 až < 1/100), vzácné (> 1/10 000 až < 1/1 000) a velmi vzácné (< 1/10 000), není známo (z dostupných údajů nelze určit).
|
Třída orgánových systémů |
Nežádoucí účinek |
Frekvence |
|
Infekce a infestace |
kandidóza, stafylokokové infekce |
vzácné |
|
Poruchy krve a lymfatického systému |
febrilní neutropenie, anemie |
méně časté |
|
Poruchy imunitního systému |
hypersenzitivní reakce včetně anafylaktických reakcí |
není známo |
|
Poruchy metabolismu a výživy |
snížení chuti k jídlu |
časté |
|
polydipsie |
vzácné | |
|
Psychiatrické poruchy |
méně časté | |
|
dezorientace, euforická nálada |
vzácné | |
|
Poruchy nervového systému |
časté | |
|
závrať, somnolence |
méně časté | |
|
kognitivní poruchy, letargie, poruchy vnímání chutí |
vzácné | |
|
Poruchy oka |
konjunktivitida |
vzácné |
|
Poruchy ucha a labyrintu |
tinnitus |
vzácné |
|
Srdeční poruchy |
palpitace |
méně časté |
|
bradykardie, kardiovaskulární porucha |
vzácné | |
|
Cévní poruchy |
návaly horka/zarudnutí |
méně časté |
|
Respirační, hrudní |
škytavka |
časté |
|
Třída orgánových systémů |
Nežádoucí účinek |
Frekvence |
|
a me diastinální poruchy |
bolest v orofaryngu, kýchání, kašel, postnazální zatékání hlenu (postnasal drip), podráždění hrdla |
vzácné |
|
Gastrointestinální poruchy |
zácpa, dyspepsie |
časté |
|
říhání, nauzeaT, zvraceníT, gastroesofageální refluxní choroba, bolest břicha, sucho v ústech, flatulence |
méně časté | |
|
perforace duodenálního vředu, stomatitida, abdominální distenze, tvrdá stolice, neutropenická kolitida |
vzácné | |
|
Poruchy kůže a podkožní tkáně |
vyrážka, akné |
méně časté |
|
fotosenzitivní reakce, hyperhidróza, seborea, kožní léze, svědivá vyrážka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza |
vzácné | |
|
svědění, kopřivka |
není známo | |
|
Poruchy svalové a kosterní soustavy a pojivové tkáně |
svalová slabost, svalové spasmy |
vzácné |
|
Poruchy ledvin a močových cest |
méně časté | |
|
polakisurie |
vzácné | |
|
Celkové poruchy a reakce v místě aplikace |
časté | |
|
astenie, celkový pocit nemoci |
méně časté | |
|
edém, pocit nepohody na hrudi, poruchy chůze |
vzácné | |
|
Vyšetření |
zvýšení ALT |
časté |
|
zvýšení AST, zvýšení alkalické fosfatázy |
méně časté | |
|
pozitivní test na červené krvinky v moči, zvýšení hladiny sodíku v krvi, pokles tělesné hmostnosti, pokles počtu neutrofilů, přítomnost glukózy v moči, zvýšená tvorba moči |
vzácné |
fNausea a zvracení byly parametry účinnosti v prvních 5-ti dnech následujících po chemoterapeutické léčbě a byly hlášeny jako nežádoucí příhody pouze poté.
Popis vybraných nežádoucích účinků
Profily nežádoucích reakcí u dospělých v prodloužených studiích s vysoce nebo středně emetogenní chemoterapií s opakovanými až 6 dalšími cykly chemoterapie byly celkově podobné jako profily pozorované v 1. cyklu.
V další aktivním komparátorem kontrolované klinické studii u 1 169 pacientů léčených aprepitantem a vysoce emetogenní terapií byl profil nežádoucích účinků obecně podobný profilu, který byl pozorovaný v ostatních studiích s vysoce emetogenní terapií s aprepitantem.
U dospělých pacientů léčených aprepitantem pro pooperační nevolnost a zvracení (PONV) byly pozorovány další nežádoucí reakce s větším výskytem než u ondansetronu: bolest horní poloviny břicha, abnormální střevní zvuky, zácpa*, dysartrie, dušnost, hypoestézie, insomnie, mióza, nevolnost, smyslové poruchy, žaludeční dyskomfort, subileus*, snížená ostrost vidění, sípot.
*Hlášeno u pacientů užívajících vyšší dávky aprepitantu.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V případě předávkování je nutno EMEND vysadit a zajistit obecnou podpůrnou léčbu a sledování pacienta. Vzhledem k antiemetickému účinku aprepitantu může snaha o vyvolání zvracení pomocí léčivých přípravků selhat.
Aprepitant nelze odstranit hemodialýzou.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antiemetika a antinauzeancia, ATC kód: A04AD12
Aprepitant je selektivní antagonista s vysokou afinitou pro receptory neurokininu 1 (NKi) lidské substance P.
Třídenní režim aprepitantu u dospělých
Ve 2 randomizovaných, dvojitě zaslepených studiích zahrnujících celkem 1 094 pacientů léčených chemoterapií obsahující cisplatinu > 70 mg/m2, byl aprepitant v kombinaci s léčebným režimem ondansetron/dexamethason (viz bod 4.2) srovnáván se standardním režimem (placebo plus ondansetron 32 mg aplikovaný intravenózně 1. den plus dexamethason 20 mg perorálně podaný 1. den a 8 mg perorálně dvakrát denně 2. až 4. den). I když se v klinických hodnoceních používala 32mg intravenózní dávka ondansetronu, nejedná se již o doporučenou dávku. Ohledně příslušného dávkování zvoleného antagonisty 5-HT3 viz informace o daném přípravku.
Účinnost se hodnotila pomocí následujícího souhrnného parametru: úplná odpověď (definovaná jako žádné emetické epizody, bez použití „záchranné“ terapie) primárně v 1. cyklu. Výsledky se hodnotily v každé studii jednotlivě a pak pro tyto 2 studie dohromady.
Přehled hlavních výsledků studií z kombinované analýzy je uveden v tabulce 1.
Tabulka 1
Procenta dospělých pacientů léčených vysoce emetogenní chemoterapií s odpovědí na léčbu podle
|
léčebných skupin a fáze |
- 1. cyklus | ||
|
SOUHRNNÝ |
Režim |
Standardní |
Rozdíly* |
|
PARAMETR |
s aprepitantem |
terapie | |
|
(N = 521) ^ |
(N = 524)T |
% (95% CI) | |
|
% |
% |
Úplná odpověď (bez zvracení a „záchranné“ terapie)
|
Celkem (0-120 hodin) 0-24 hodin 25-120 hodin |
67,7 86,0 71,5 |
47,8 73.2 51.2 |
19,9 12,7 20,3 |
(14,0, 25,8) (7,9, 17,6) (14,5, 26,1) |
|
JEDNOTLIVÁ KRITÉRIA | ||||
|
Bez zvracení (žádné emetické epizody bez ohledu |
na použití „záchranné“ terapie) | |||
|
Celkem (0-120 hodin) |
71,9 |
49,7 |
22,2 |
(16,4, 28,0) |
|
0-24 hodin |
86,8 |
74,0 |
12,7 |
(8,0, 17,5) |
|
25-120 hodin |
76,2 |
53,5 |
22,6 |
(17,0, 28,2) |
|
Bez významné nevolnosti (maximálně VAS <25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
72,1 |
64,9 |
7,2 |
(1,6, 12,8) |
|
25-120 hodin |
74,0 |
66,9 |
7,1 |
(1,5, 12,6) |
* Intervaly spolehlivosti (CI) byly vypočítány bez korekce pro pohlaví a současnou chemoterapii, které byly zahrnuty do primární analýzy míry pravděpodobnosti a logistických modelů. t Jeden pacient v režimu pouze s aprepitantem měl data v akutní fázi a byl vyřazen z celkové a prodloužené fáze analýzy: jeden pacient pouze ve standardním režimu měl pouze data v prodloužené fázi a byl vyřazen z celkové a akutní fáze analýzy.
Odhad doby první epizody zvracení v kombinované analýze je zobrazen vynesením hodnot metodou Kaplana-Meiera v grafu 1.
Graf 1
Procento dospělých pacientů léčených vysoce emetogenní chemoterapií, u nichž nedošlo k epizodě zvracení - 1. cyklus

0 12 24 36 48 60 72 84 96 108 120
Čas (v hodinách)
Statisticky významné rozdíly v účinnosti byly pozorovány v každé z těchto 2 jednotlivých studií.
V těchto stejných 2 klinických studiích pokračovalo 851 dospělých pacientů v prodloužené studii
s opakovanými až 5 dalšími cykly chemoterapie. Účinnost režimu s aprepitantem zůstala během všech cyklů zřejmě zachována.
V randomizované, dvojitě zaslepené studii s celkovým počtem 866 dospělých pacientů (864 žen,
2 muži) léčených chemoterapií, která zahrnovala cyklofosfamid 750-1 500 mg/m2; nebo cyklofosfamid 500-1 500 mg/m2 a doxorubicin (< 60 mg/m2) nebo epirubicin (< 100 mg/m2), byl aprepitant v kombinaci s režimem obsahujícím ondansetron/dexamethason (viz bod 4.2) srovnáván se standardní terapií (placebo plus ondansetron 8 mg perorálně (dvakrát 1. den a každých 12 hodin 2. a 3. den) plus dexamethason 20 mg perorálně 1. den).
Účinnost se hodnotila pomocí souhrnného parametru: úplná odpověď (definovaná jako žádné emetické epizody a nepoužití záchranné terapie) primárně během 1. cyklu.
Přehled hlavních výsledků studie je uveden v Tabulce 2.
Tabulka 2
Procenta dospělých pacientů odpovídajících na léčbu podle léčebné skupiny a fáze - 1. cyklus
středně emetogenní chemoterapie
|
SOUHRNNÝ |
Režim |
Standardní |
Rozdíly* |
|
PARAMETR |
s aprepitante |
terapie | |
|
m (N = 433)t % |
(N = 424) % |
% (95 % CI) |
|
Úplná odpověď (bez zvracení a |
„záchranné“ terapie) | |||
|
Celkem (0-120 hodin) |
50,8 |
42,5 |
8,3 |
(1,6, 15,0) |
|
0-24 hodin |
75,7 |
69,0 |
6,7 |
(0,7, 12,7) |
|
25-120 hodin |
55,4 |
49,1 |
6,3 |
(-0,4, 13,0) |
|
jednotlivá kritéria | ||||
|
Bez zvracení (žádné emetické epizody bez ohledu na |
použití „záchranné“ |
terapie) | ||
|
Celkem (0-120 hodin) |
75,7 |
58,7 |
17,0 |
(10,8, 23,2) |
|
0-24 hodin |
87,5 |
77,3 |
10,2 |
(5,1, 15,3) |
|
25-120 hodin |
80,8 |
69,1 |
11,7 |
(5,9, 17,5) |
|
Bez významné nevolnosti (maximálně VAS <25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
60,9 |
55,7 |
5,3 |
(-1,3, 11,9) |
|
0-24 hodin |
79,5 |
78,3 |
1,3 |
(-4,2, 6,8) |
|
25-120 hodin |
65,3 |
61,5 |
3,9 |
(-2,6, 10,3) |
* Intervaly spolehlivosti byly vypočítány bez korekce na věkovou kategorii (<55 let, >55 let) a skupiny hodnotících lékařů, které byly zahrnuty do primární analýzy míry pravděpodobnosti a logistických modelů.
t Jeden pacient v režimu pouze s aprepitantem měl data v akutní fázi a byl vyřazen z celkové a prodloužené fáze analýzy.
Ve stejné klinické studii pokračovalo 744 dospělých pacientů v prodloužené studii s opakovanými až 3 dalšími cykly chemoterapie. Účinnost režimu s aprepitantem zůstala během všech cyklů zřejmě zachována.
Ve druhé multicentrické, randomizované, dvojitě zaslepené studii s paralelní skupinou byl režim s aprepitantem porovnáván se standardní léčbou u 848 dospělých pacientů (652 žen, 196 mužů), kterým byl podáván chemoterapeutický režim, který zahrnoval jakékoli intravenózní dávky oxaliplatiny, karboplatiny, epirubicinu, idarubicinu, ifosfamidu, irinotekanu, daunorubicinu, doxorubicinu; cyklofosfamid intravenózně (< 1 500 mg/m2) nebo cytarabin intravenózně (>1 g/m2). Pacientům, kterým byl podáván režim s aprepitantem, byla chemoterapie podávána na různé typy nádorů, včetně 52 % s karcinomem prsu, 21 % s gastrointestinálními karcinomy včetně kolorektálního karcinomu, 13 % s karcinomem plic a 6 % s gynekologickými karcinomy. Režim s aprepitantem v kombinaci s režimem ondansetron/dexamethason (viz bod 4.2) byl porovnáván se standardní léčbou
(placebo v kombinaci s ondansetronem 8 mg perorálně (dvakrát 1. den a každých 12 hodin 2. a 3. den) plus dexamethason 20 mg perorálně 1. den).
Účinnost byla založena na vyhodnocení následujících primárních a klíčových sekundárních cílových parametrů účinnosti: žádné zvracení za celou dobu (0 až 120 hodin po chemoterapii), vyhodnocení bezpečnosti a snášenlivosti režimu s aprepitantem při chemoterapií vyvolané nevolnosti a zvracení (CINV) a úplná odpověď (definovaná jako žádné zvracení a žádné použití záchranné léčby) za celou dobu (0 až 120 hodin po chemoterapii). Navíc byla sledována jako exploratorní cílový parametr účinnosti, „Žádná významná nauzea“ (No Significant Nausea) za celou dobu (0 až 120 hodin po chemoterapii), jak v akutní, tak i prodloužené fázi formou post-hoc analýzy.
Souhrn klíčových výsledků studie je uveden v Tabulce 3.
Tabulka 3
Procento dospělých pacientů odpovídajících na léčbu podle léčebné skupiny a fáze pro studii 2 -
cyklus 1
středně emetogenní chemoterapie
Režim Standardní Rozdíly*
s aprepi- léčba
tantem (N = 406) % (95% CI)
(N = 425) %
%
Úplná odpověď (žádné zvracení a žádná záchranná léčba)
|
Celkem (0-120 hodin) 0-24 hodin 25-120 hodin |
68.7 89,2 70.8 |
56.3 80.3 60,9 |
12,4 8.9 9.9 |
(5,9, 18,9) (4,0, 13,8) (3,5, 16,3) |
|
Žádná emeze (žádné emetické epizody bez ohledu |
na použití záchranné léčby) | |||
|
Celkem (0-120 hodin) |
76,2 |
62,1 |
14,1 |
(7,9, 20,3) |
|
0-24 hodin |
92,0 |
83,7 |
8,3 |
(3,9, 12,7) |
|
25-120 hodin |
77,9 |
66,8 |
11,1 |
(5,1, 17,1) |
|
Žádná významná nauzea (maximální VAS <25 mm na stupnici 0-100 mm) | ||||
|
Celkem (0-120 hodin) |
73,6 |
66,4 |
7,2 |
(1,0, 13,4) |
|
0-24 hodin |
90,9 |
86,3 |
4,6 |
(0,2, 9,0) |
|
25-120 hodin |
74,9 |
69,5 |
5,4 |
(-0,7, 11,5) |
*Intervaly spolehlivosti byly vypočteny bez úpravy podle pohlaví a oblasti, které byly zařazeny v primární analýze za použití logistických modelů.
Přínos kombinované terapie s aprepitantem u celé hodnocené populace byl dán zejména výsledky pozorovanými u pacientů se slabou kontrolou při standardním režimu, jako jsou ženy, i když výsledky byly numericky lepší bez ohledu na věk, typ tumoru nebo pohlaví. Úplné odpovědi bylo dosaženo u 209/324 (65 %) v případě režimu s aprepitantem a u 161/320 (50 %) při standardní terapii u žen a u mužů šlo v případě režimu s aprepitantem u 83/101 (82 %) a při standardní terapii u 68/87 (78 %).
Pediatrická populace
V randomizované, dvojitě zaslepené klinické studii, kontrolované aktivním komparátorem, která zahrnovala 302 dětí a dospívajících (ve věku 6 měsíců až 17 let), kterým se podávala středně silně nebo vysoce emetogenní chemoterapie, se režim s aprepitantem porovnával s kontrolním režimem prevence CINV. Účinnost režimu s aprepitantem byla hodnocena při jednom cyklu (cyklus 1). Pacienti měli v následných cyklech (volitelné cykly 2 až 6) možnost dostávat aprepitant v otevřeném uspořádání; v těchto volitelných cyklech však účinnost hodnocena nebyla. Režim s aprepitantem u dospívajících ve věku 12 až 17 let (n=47) sestával z přípravku EMEND tobolky 125 mg podávaného perorálně 1. den a z 80 mg/den 2. a 3. den v kombinaci s ondansetronem 1. den. Režim s aprepitantem u dětí ve věku 6 měsíců až méně než 12 let (n=105) sestával z přípravku EMEND prášek pro perorální
suspenzi podávaného v dávce 3,0 mg/kg (do dávky 125 mg) perorálně 1. den a v dávce 2,0 mg/kg (do dávky 80 mg) perorálně 2. a 3. den v kombinaci s ondansetronem 1. den. Kontrolní režim u dospívajících ve věku 12 až 17 let (n=48) a dětí ve věku 6 měsíců až méně než 2 let (n=102) sestával z placeba místo aprepitantu 1., 2. a 3. den v kombinaci s ondansetronem 1. den. Přípravek EMEND nebo placebo a ondansetron se podávaly 1 hodinu, respektive 30 minut před zahájením chemoterapie. Jako součást antiemetického režimu pro pediatrické pacienty byl u obou věkových skupin povolen intravenózní dexamethason, a to podle rozhodnutí lékaře. U pediatrických pacientů léčených aprepitantem bylo požadováno snížení (50%) dávky dexamethasonu. U pediatrických pacientů léčených kontrolním režimem nebylo snížení dávky požadováno. Z pediatrických pacientů používalo v
1. cyklu ve skupině léčené režimem s aprepitantem dexamethason 29 % zúčastněných a ve skupině s kontrolním režimem 28 % zúčastněných.
Antiemetická aktivita přípravku EMEND byla hodnocena během 5denního (120 hodin) období po zahájení chemoterapie 1. den. Primárními kritérii hodnocení byla úplná odpověď v pozdní fázi (25 až 120 hodin po zahájení chemoterapie) v 1. cyklu. Souhrn klíčových výsledků studie je uveden v Tabulce 4.
Tabulka 4
Počet (%) pediatrických pacientů s úplnou odpovědí a bez zvracení podle léčebné skupiny a fáze - 1.
cyklus (populace všech zařazených pacientů)
|
Režim s aprepitanten n/m (%) |
Kontrolní režim n/m (%) | |
|
PRIMÁRNÍ KRITÉRIUM HODNOCENÍ | ||
|
Úplná odpověď * - pozdní fáze |
77/152 (50,7)t |
39/150 (26,0) |
|
DALŠÍ PŘEDEM DEFINOVANÁ KRITÉRIA HODNOCENÍ | ||
|
Úplná odpověď * - akutní fáze |
101/152 (66,4); |
78/150 (52,0) |
|
Úplná odpověď - úplná fáze |
61/152 (40,1)t |
30/150 (20,0) |
|
i* Bez zvracení § - úplná fáze |
71/152 (46,7)t |
32/150 (21,3) |
|
*Úplná odpověď = žádné zvracení ani říhání nebo zvedání žaludku naprázc záchranné medikace. Tp < 0,01 v porovnání s kontrolním režimem *p < 0,05 v porovnání s kontrolním režimem §Žádné zvracení = žádné zvracení ani říhání nebo zvedání žaludku naprázdno n/m = počet pacientů s požadovanou odpovědí/počet pacientů zařazených v daný o Akutní fáze: 0 až 24 hodin po zahájení chemoterapie. Pozdní fáze: 25 až 120 hodin po zahájení chemoterapie. Úplná fáze: 0 až 120 hodin po zahájení chemoterapie. |
no a žádné použití kamžik. | |
Odhadovaná doba do prvního zvracení po zahájení léčby chemoterapií byla u režimů s aprepitantem delší (odhadovaný medián času do prvního zvracení byl 94,5 hodiny) v porovnání se skupinou s kontrolním režimem (odhadovaný medián času do prvního zvracení byl 26,0 hodiny), jak je uvedeno Kaplan-Meierovými křivkami na Grafu 2.
Dob a do první epizody zvracení od začátku podávání chemoterapie pediatrickým pacientům v celkové
fázi 1. cyklu (populace všech zařazených pacientů)

Analýza účinnosti u subpopulací v 1. cyklu prokázala, že režim s aprepitantem poskytoval lepší kontrolu než kontrolní režim, pokud jde o kritérium hodnocení úplná odpověď, a to bez ohledu na věkovou kategorii, pohlaví, podání dexamethasonu k antiemetické profylaxi a emetogenitu chemoterapie.
5.2 Farmakokinetické vlastnosti
Farmakokinetika aprepitantu má nelineární charakter. Clearance i absolutní biologická dostupnost se se zvyšováním dávky snižují.
Absorpce
Střední hodnota biologické dostupnosti perorálně podaného aprepitantu činí 67 % u 80mg tobolky a 59 % u 125mg tobolky. Průměrné maximální plazmatické koncentrace (Cmax) aprepitantu byly dosaženy přibližně po 4 hodinách (tmax). Perorální podání tobolky spolu se standardní snídaní s kalorickou hodnotou přibližně 800 kcal vedlo až k 40% zvýšení hodnoty AUC aprepitantu. Toto zvýšení se nepovažuje za klinicky významné.
Farmakokinetika aprepitantu má nelineární charakter v celém rozsahu klinických dávek. U mladých zdravých dospělých jedinců bylo zvýšení hodnoty AUC0-<X) o 26 % větší, než byla proporcionální dávka mezi jednotlivými 80mg a 125mg dávkami podanými v nasyceném stavu.
Po perorálním podání jedné 125mg dávky přípravku EMEND 1. den a dávky 80 mg jednou denně
2. a 3. den dosáhla hodnota AUC0-24hod. (střední hodnota ± standardní odchylka) 19,6 ± 2,5 pg • hod/ml 1. den a 21,2 ± 6,3 pg • hod/ml 3. den. Hodnota Cmax činila 1. den 1,6 ± 0,36 pg/ml a 3. den 1,4 ± 0,22 pg/ml.
Distribuce
Aprepitant se ve vysoké míře váže na proteiny, se střední hodnotou 97 %. Geometrická střední hodnota zdánlivého distribučního objemu v rovnovážném stavu (Vdss) je u člověka přibližně 66 litrů.
Biotransformace
Aprepitant prochází rozsáhlou biotransformací. U zdravých mladých dospělých jedinců vykazuje aprepitant po dobu 72 hodin po jednorázové intravenózní aplikaci 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, v plazmě přibližně 19 % radioaktivity, což ukazuje na značnou přítomnost metabolitů v plazmě. V lidské plazmě bylo zjištěno dvanáct metabolitů aprepitantu. Metabolismus aprepitantu probíhá ve velké míře cestou oxidace v morfolinovém kruhu a jeho postranních řetězcích a výsledné metabolity jsou pouze slabě aktivní. In vitro studie s lidskými jaterními mikrozómy ukázaly, že aprepitant se biotransformuje primárně cestou CYP3A4, případně s malým podílem CYP1A2 a CYP2C19.
Aprepitant se nevylučuje močí v nezměněné podobě. Metabolity se vylučují močí a žlučí ve stolici.
Po jednorázově intravenózně aplikované dávce 100 mg [14C]-fosaprepitantu, což je proléčivo aprepitantu, zdravým jedincům bylo 57 % radioaktivity zjištěno v moči a 45 % ve stolici.
Plazmatická clearance aprepitantu závisí na dávce, se zvyšující se dávkou se snižuje a v rozmezí terapeutických dávek se pohybovala přibližně na hodnotách od 60 do 72 ml/min. Terminální poločas se pohyboval v rozmezí přibližně od 9 do 13 hodin.
Farmakokinetika u speciálních populací
Pohlaví: Po perorálním podání jediné 125mg dávky aprepitantu byla hodnota Cmax aprepitantu u žen ve srovnání s muži o 16 % vyšší. Poločas aprepitantu je u žen ve srovnání s muži o 25 % nižší a k dosažení tmax dochází zhruba ve stejnou dobu. Tyto rozdíly nejsou považovány za klinicky významné. Dávku přípravku EMEND není nutno podle pohlaví pacienta nijak upravovat.
Porucha funkce jater: Mírná porucha funkce jater (Childovo-Pughovo hodnocení, třída A) farmakokinetiku aprepitantu v klinicky významné míře neovlivňuje. U pacientů s mírnou poruchou funkce jater není třeba dávkování nijak upravovat. Z dostupných dat nelze činit žádné závěry ohledně vlivu středně těžké poruchy funkce jater (Childovo-Pughovo hodnocení, třída B) na farmakokinetiku aprepitantu. K dispozici nejsou žádné klinické ani farmakokinetické údaje od pacientů s těžkou poruchou funkce jater (Childovo-Pughovo hodnocení, třída C).
Porucha funkce ledvin: Pacientům s těžkou poruchou funkce ledvin (CrCl < 30 ml/min) a pacientům s terminálním renálním onemocněním (end stage renal disease, ESRD) s potřebou hemodialýzy byla podána jednorázová 240mg dávka aprepitantu.
U pacientů s těžkou poruchou funkce ledvin se hodnota AUC0_W celkového aprepitantu (nevázaného i vázaného na proteiny) ve srovnání se zdravými jedinci snížila o 21 % a hodnota Cmax se snížila o 32 %. U pacientů s ESRD podstupujících hemodialýzu se hodnota AUC0.OT celkového aprepitantu snížila o 42 % a hodnota Cmax se snížila o 32 %. Vzhledem k mírnému poklesu vazby aprepitantu na proteiny u pacientů s renálním onemocněním nebyla hodnota AUC farmakologicky aktivní části nenavázaného léku u pacientů s poruchou funkce ledvin ve srovnání se zdravými jedinci významně ovlivněna. Hemodialýza prováděná 4 nebo 48 hodin po podání dávky neměla na farmakokinetiku aprepitantu významný účinek; v dialyzátu bylo zjištěno méně než 0,2 % dávky.
U pacientů s poruchou funkce ledvin ani u pacientů s ESRD podstupujících hemodialýzu není zapotřebí dávkování přípravku EMEND nijak upravovat.
Pediatrická populace: V rámci 3denního režimu se u většiny pacientů dávkováním aprepitantu v tobolkách (125/80/80 mg) u dospívajících pacientů (ve věku 12 až 17 let) první den dosáhlo AUC0_24hod nad 17 ^g^hod/ml s koncentracemi (Cmin) na konci 2. a 3. dne nad 0,4 ^g/ml. Medián maximálních plazmatických koncentrací (Cmax) byl 1. den přibližně 1,3 ^g/ml a tato koncentrace se objevila za přibližně 4 hodiny. V rámci 3denního režimu se u většiny pacientů dávkováním aprepitantu v prášku pro přípravku perorální suspenze (3/2/2 mg/kg) u pacientů ve věku 6 měsíců až méně než 12 let první den dosáhlo AUC0.24hod nad 17 ^g^hod/ml s koncentracemi (Cmin) na konci 2. a
3. dne nad 0,1 ^g/ml. Medián maximálních plazmatických koncentrací (Cmax) byl první den přibližně
1,2 (ig/ml a tato koncentrace se objevila za 5 až 7 hodin.
Populační farmakokinetická analýza aprepitantu u pediatrických pacientů (ve věku 6 měsíců až 17 let) naznačuje, že pohlaví a rasa na farmakokinetiku aprepitantu klinicky významný vliv nemají.
Vztah mezi koncentrací a účinkem
Studie využívající pozitronovou emisní tomografii (PET) s použitím vysoce specifického značení receptorů NK1 u zdravých mladých mužů ukázaly, že aprepitant prostupuje do mozku a obsazuje receptory NK v závislosti na dávce a koncentraci v plazmě. Předpokládá se, že plazmatické
koncentrace aprepitantu dosažené při 3denním léčebném režimu s přípravkem EMEND u dospělých zajišťují více než 95% obsazení receptorů NK1 v mozku.
5.3 Předklinické údaje vztahující se k bezpečnosti
Předklinické údaje získané na základě konvenčních studií toxicity, genotoxicity, hodnocení kancerogenního potenciálu, reprodukční a vývojové toxicity po jednorázovém a opakovaném podání přípravku neodhalily žádné zvláštní riziko pro člověka. Je však nutno mít na paměti, že systémová expozice u hlodavců byla podobná nebo dokonce nižší, než byla terapeutická expozice u člověka při dávce 125 mg/80 mg. I když nebyly v reprodukčních studiích s expozičními hodnotami pro člověka zjištěny žádné nežádoucí účinky, nelze zejména na základě hodnot expozice použitých u zvířat dostatečně posoudit riziko pro člověka.
Ve studii juvenilní toxicity na potkanech, kterým se podával aprepitant od 10. do 63. postnatálního dne, docházelo u samic k časnému vaginálnímu otevírání od dávek 250 mg/kg dvakrát denně a u samců k opožděné prepuciální separaci, a to od dávek 10 mg/kg dvakrát denně. Nebyl zde žádný odstup od klinicky relevantní expozice. Nedošlo k žádným účinkům souvisejícím s léčbou ohledně páření, fertility či embryonálního/fetálního vývoje a nevyskytly se žádné patologické změny reprodukčních orgánů. Ve studii juvenilní toxicity na psech ošetřovaných od 14. do 42. postnatálního dne byla u samců při dávce 6 mg/kg/den pozorována snížená hmotnost varlat a velikost Leydigových buněk a u samic při dávkách od 4 mg/kg/den byla pozorována zvýšená hmotnost dělohy, hypertrofie dělohy a krčku a otok vaginálních tkání. Nebyl zde žádný odstup od klinicky relevantní expozice aprepitantu. Při krátkodobé léčbě podle doporučeného dávkovacího režimu se klinická relevance těchto zjištění nepovažuje za pravděpodobnou.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Hyprolóza (E 463)
Natrium-lauryl-sulfát Sacharóza Laktóza (bezvodá)
Červený oxid železitý (E 172)
Natrium-stearyl-fumarát (E 485)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
Neotevřený sáček: 2 roky
Po rekonstituci: Perorální suspenze může být uchovávána při pokojové teplotě po dobu až 3 hodiny (při teplotě do 30 °C). Také může být uchovávána v chladničce po dobu až 72 hodin (při teplotě 2 °C -8 °C).
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání. Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Podmínky uchovávání po rekonstituci léčivého přípravku naleznete v bodě 6.3.
6.5 Druh obalu a obsah balení
PET/Al/LLDPE sáčky.
Krabička na jedno použití.
Jedna krabička obsahuje jeden sáček s práškem pro perorální suspenzi, jeden 1ml a jeden 5ml perorální dávkovač (polypropylenový se silikonovým o-kroužkem), jeden uzávěr a jednu mísicí nádobku (polypropylenovou).
6.6 Zvláštní opatření pro likvidaci přípravku
Obsah jednoho jednorázového sáčku se suspenduje v 4,6 ml vody, čímž se získá konečná koncentrace 25 mg na mililitr.
• Více informací o přípravě a podávání suspenze naleznete v příbalové informaci a v instrukcích pro přípravu perorální suspenze pro zdravotnické pracovníky.
• K odměření 4,6 ml vody použijte 5ml perorální dávkovač, který je přiložen k mísicí nádobce.
• Celý obsah sáčku vsypte do 4,6 ml vody a promíchejte.
• Po promíchání odměřte pomocí perorální dávkovače doporučený objem (dávku) suspenze
a perorálně podejte dávku. Na základě dávky zvolte perorální dávkovač. Zvolte 1ml perorální dávkovač je-li dávka 1 ml a méně a 5ml perorální dávkovač je-li dávka větší než 1 ml. Dávku podejte perorálně. Pokud není dávka podána bezprostředně po odměření, může být naplněný perorální dávkovač uchován v chladu (při teplotě 2 °C - 8 °C) po dobu až 72 hodin před použitím.
• Perorální suspenze může být před podáním uchovávána při pokojové teplotě (při teplotě do 30 °C).
• Všechnu zbývající suspenzi a odpadní materiál zlikvidujte.
Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd.
Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
8. REGISTRAČNÍ ČÍSLO
EU/1/03/262/011
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 11. listopadu 2003 Datum prodloužení registrace: 22. září 2008
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ/VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ/ VÝROBCI ODPOVĚDNÍ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného/výrobců odpovědných za propouštění šarží
Merck Sharp & Dohme B.V.
Waarderweg 39 2031 BNHaarlem Nizozemsko
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis.
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
VNĚJŠÍ OBAL - STANDARDNÍ BALENÍ (KRABIČKA)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 40 mg tvrdé tobolky Aprepitantum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje aprepitantum 40 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sacharózu. Další údaje viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 tvrdá tobolka 5x1 tvrdá tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/262/007 1 tvrdá tobolka EU/1/03/262/008 5 x 1 tvrdá tobolka
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
EMEND 40 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH A STRIPECH TEXT NA BLISTRU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 40 mg tvrdé tobolky Aprepitantum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
MSD
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
EMEND 80 mg tvrdé tobolky Aprepitantum
Jedna tvrdá tobolka obsahuje aprepitantum 80 mg.
Obsahuje sacharózu. Další údaje viz příbalová informace.
1 tvrdá tobolka
Balení pro 2denní léčbu obsahující: 2 x 80mg tvrdou tobolku 5x1 tvrdá tobolka
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/262/001 1 tvrdá tobolka EU/1/03/262/002 2 x 1 tvrdá tobolka EU/1/03/262/003 5 x 1 tvrdá tobolka
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
EMEND 80 mg
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU (OBSAHUJÍCÍM 2 TVRDÉ TOBOLKY 80 MG) VNITŘNÍ OBAL - TROJDÍLNÁ SKLÁDAČKA - balení pro 2denní léčbu_
EMEND 80 mg tvrdé tobolky Aprepitantum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje aprepitantum 80 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sacharózu. Další údaje viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
Balení pro 2denní léčbu obsahující: 2 x 80mg tvrdou tobolku
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Perorální podání.
Před použitím si přečtěte příbalovou informaci.
Další informace o způsobu užití přípravku EMEND naleznete v příbalové informaci.
KDY a JAK užívat EMEND
Váš lékař Vám předepsal EMEND, antiemetikum, lék k předcházení nevolnosti a zvracení spojených s chemoterapií.
JAK:
EMEND 80 mg tvrdé tobolky se užívají jen jednou denně během 2 následujících dnů.
Tobolky EMEND se mohou užívat s jídlem nebo i bez něj.
Nevyjímejte najednou všechny tobolky.
Tobolku vyjměte protlačením z této strany.
Začátek léčby KDY:
Jednu EMEND 80mg tobolku užívejte každé ráno. Začněte v den následující po chemoterapii.
Den 1 Den 2
EMEND 80mg tobolka
Vzhledem k tomu, že nevolnost a zvracení se mohu objevit ve dnech následujících po Vaší chemoterapii, je důležité užívat EMEND během 2 následujících dnů tak, jak Vám předepsal Váš lékař.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/262/002
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH TEXT NA BLISTRU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 80 mg tvrdé tobolky Aprepitantum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
MSD
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
VNĚJŠÍ OBAL - STANDARDNÍ BALENÍ (SKLÁDAČKA)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 125 mg tvrdé tobolky Aprepitantum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje aprepitantum 125 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sacharózu. Další údaje viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 tvrdá tobolka 5x1 tvrdá tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/262/004 1 tvrdá tobolka EU/1/03/262/005 5 x 1 tvrdá tobolka
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
EMEND 125 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH TEXT NA BLISTRU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 125 mg tvrdé tobolky Aprepitantum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
MSD
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
ÚDAJE UVÁDĚNÉ NA VNĚJŠÍM OBALU
VNĚJŠÍ OBAL - STANDARDNÍ BALENÍ (PAPÍROVÁ KRABIČKA)
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 165 mg tvrdé tobolky Aprepitantum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tvrdá tobolka obsahuje 165 mg aprepitantum.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje sacharózu. Další údaje jsou uvedeny v příbalové informaci.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
1 tvrdá tobolka 6x1 tvrdá tobolka
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/262/009 1 tvrdá tobolka EU/1/03/262/010 6 x 1 tvrdá tobolka
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
EMEND 165 mg
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH TEXT NA BLISTRU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 165 mg tvrdé tobolky Aprepitantum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
MSD
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. JINÉ
EMEND 125 mg tvrdá tobolka EMEND 80 mg tvrdé tobolky Aprepitantum
Jedna 125mg tvrdá tobolka obsahuje aprepitantum 125 mg. Jedna 80mg tvrdá tobolka obsahuje aprepitantum 80 mg.
Obsahuje sacharózu. Další údaje viz příbalová informace.
Balení pro 3denní léčbu obsahující:
1 x 125mg tvrdá tobolka a
2 x 80mg tvrdá tobolka
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/262/006
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
EMEND 125 mg/80 mg
ÚDAJE UVÁDĚNÉ NA VNITŘNÍM OBALU (OBSAHUJÍCÍM 1 TVRDOU TOBOLKU 125 MG A 2 TVRDÉ TOBOLKY 80 MG)
VNITŘNÍ OBAL - TROJDÍLNÁ SKLÁDAČKA - balení pro 3denní léčbu_
EMEND 125 mg tvrdá tobolka EMEND 80 mg tvrdé tobolky Aprepitantum
Jedna 125mg tvrdá tobolka obsahuje aprepitantum 125 mg. Jedna 80mg tvrdá tobolka obsahuje aprepitantum 80 mg.
Obsahuje sacharózu. Další údaje viz příbalová informace.
Balení pro 3denní léčbu obsahující:
1 x 125mg tvrdá tobolka a
2 x 80mg tvrdá tobolka
Perorální podání.
Před použitím si prosím přečtěte příbalovou informaci.
Další informace o způsobu užití přípravku EMEND naleznete v příbalové informaci.
KDY a JAK užívat EMEND
Váš lékař Vám předepsal EMEND, antiemetikum, lék k předcházení nevolnosti a zvracení spojených s chemoterapií.
JAK:
EMEND se užívá jen jednou denně během 3 následujících dnů.
Tobolky EMEND se mohou užívat s jídlem nebo i bez něj.
Nevyjímejte najednou všechny tobolky.
Tobolku vyjměte protlačením z této strany.
Začátek léčby KDY:
Jednu EMEND 125mg tobolku užijte ústy 1 hodinu PŘED začátkem Vaší chemoterapie.
1. den
EMEND 125mg tobolka KDY:
Jednu EMEND 80mg tobolku užívejte každé ráno v následujících dvou dnech.
2. den
3. den
EMEND 80mg tobolka
Vzhledem k tomu, že nevolnost a zvracení se mohu objevit ve dnech následujících po Vaší chemoterapii, je důležité užívat EMEND během 3 následujících dnů tak, jak Vám předepsal Váš lékař.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z TAKOVÝCH LÉČIVÝCH PŘÍPRAVKŮ, POKUD JE TO VHODNÉ
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
EU/1/03/262/006
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
EMEND 125 mg prášek pro perorální suspenzi aprepitantum
Pro děti ve věku 6 měsíců až méně než 12 let
Jeden sáček obsahuje aprepitantum 125 mg. Po rekonstituci obsahuje 1 ml perorální suspenze aprepitantum 25 mg.
Obsahuje sacharózu a laktózu. Další údaje viz příbalová informace.
Prášek pro perorální suspenzi
Jeden sáček, dva perorální dávkovače, jeden uzávěr a jedna mísicí nádobka.
Před použitím si přečtěte příbalovou informaci. Perorální podání.
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/03/262/011
13. ČÍSLO ŠARŽE
Č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
EMEND 125 mg prášek pro perorální suspenzi
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA MALÉM VNITŘNÍM OABLU Sáček přípravku EMEND 125 mg prášek pro perorální suspenzi
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
EMEND 125 mg prášek pro perorální suspenzi
aprepitantum
Perorální podání
2. ZPŮSOB PODÁNÍ
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
Lot
5. OBSAH UDANÝ JAKO HMOTNOST, OBJEM NEBO POČET
6. JINÉ
Příbalová informace: informace pro uživatele
EMEND 40 mg tvrdé tobolky
aprepitantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí
ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je EMEND a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat
3. Jak se EMEND užívá
4. Možné nežádoucí účinky
5 Jak EMEND uchovávat
6. Obsah balení a další informace
1. Co je EMEND a k čemu se používá
Přípravek EMEND obsahuje léčivou látku aprepitant, která patří do skupiny léčiv nazývaných „antagonisté receptoru neurokininu 1 (NK1)“(léky potlačující zvracení a pocit na zvracení). Mozek má zvláštní oblast, která řídí pocit na zvracení a zvracení. Přípravek EMEND funguje tak, že blokuje signály do této oblasti, a používá se u dospělých k předcházení nevolnosti a zvracení způsobených operací.
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat Neužívejte přípravek EMEND:
- jestliže jste alergický(á) na aprepitant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Před užitím přípravku EMEND se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Pokud trpíte onemocněním jater, řekněte to před léčbou přípravkem EMEND svému lékaři, protože játra hrají důležitou roli při rozkladu léku v těle. Proto Váš lékař může potřebovat sledovat stav Vašich jater.
Děti a dospívající
Nepodávejte přípravek EMEND 40 mg dětem a dospívajícím mladším 18 let, protože 40mg tobolky nebyly u této populace hodnoceny.
Další léčivé přípravky a přípravek EMEND
Přípravek EMEND může ovlivňovat účinky jiných léků.
Účinky přípravku EMEND nebo jiných léčiv mohou být ovlivněny, pokud budete přípravek EMEND užívat spolu s dalšími léky, včetně léků uvedených dále. Poraďte se prosím se svým lékařem nebo lékárníkem, pokud užíváte některý z následujících léků:
- léky k zabránění početí, které mohou zahrnovat antikoncepční pilulky, kožní náplasti, implantáty a některá nitroděložní tělíska, která uvolňují hormony, nemusí fungovat odpovídajícím způsobem, pokud se používají spolu s přípravkem EMEND. Během léčby přípravkem EMEND a až 2 měsíce po jeho použití je nutno používat jiné nebo dodatečné nehormonální formy antikoncepce,
- pimozid (používá se k léčbě psychiatrických onemocnění),
- terfenadin; astemizol (používají se proti senné rýmě a jiným alergickým stavům),
- cisaprid (používá se k léčbě trávících potíží),
- léky obsahující deriváty ergotových alkaloidů (používají se k léčbě migrén),
- rifampicin, klarithromycin, telithromycin (antibiotika používaná k léčbě infekcí),
- fenytoin (lék používaný k léčbě záchvatů),
- karbamazepin (používaný se k léčbě depresí a epilepsie),
- midazolam, fenobarbital (utišující léky nebo léky usnadňující spánek),
- třezalka tečkovaná (bylinný přípravek používaný k léčbě deprese),
- inhibitory proteázy (používané k léčbě infekcí HIV),
- nefazodon (používá se k léčbě deprese),
- ketokonazol, kromě šampónu (používá se k léčbě Cushingova syndromu - kdy tělo vytváří nadměrná množství kortizolu)
- itrakonazol, vorikonazol, posakonazol (léky proti plísním)
- kortikosteroidy (jako je dexamethason).
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Neužívejte tento přípravek během těhotenství, pokud to není nezbytně nutné. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat.
Ohledně informací týkajících se antikoncepce viz část "Další léčivé přípravky a přípravek EMEND".
Není známo, zda se přípravek EMEND vylučuje do lidského mateřského mléka; proto není doporučeno kojit během léčby tímto přípravkem. Pokud kojíte nebo hodláte kojit, je třeba, abyste tuto skutečnost svému lékaři sdělila ještě předtím, než začnete užívat tento přípravek.
Řízení dopravních prostředků a obsluha strojů
Je nutno vzít v úvahu, že u některých lidí může po užití přípravku EMEND vzniknout pocit závrati a ospalosti. Pokud máte pocit závrati a ospalosti, nesmíte po užití tohoto přípravku řídit ani obsluhovat stroje (viz „Možné nežádoucí účinky“).
Přípravek EMEND obsahuje sacharózu
Tobolky přípravku EMEND obsahují sacharózu. Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem, než začnete tento přípravek užívat.
3. Jak se EMEND užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Pokud si nejste jistý(á), poraďte se se svým lékařem nebo lékárníkem.
Doporučená perorální dávka (podaná ústy) přípravku EMEND je jedna 40mg tobolka během 3 hodin před zahájením anestézie.
Tobolku spolkněte celou a zapijte ji douškem tekutiny.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přestaňte přípravek EMEND užívat a ihned navštivte lékaře, pokud zaznamenáte kterýkoli z následujících nežádoucích účinků, které mohou být závažné a kvůli nimž můžete naléhavě potřebovat lékařské ošetření:
- kopřivka, vyrážka, svědění, potíže s dýcháním nebo polykáním (četnost není známa, z dostupných údajů ji nelze určit); jde o známky alergické reakce.
Další hlášené nežádoucí účinky jsou uvedeny dále.
Časté nežádoucí účinky (mohou postihovat až 1 z 10 lidí) jsou:
- zvýšené množství jaterních enzymů v krvi.
Méně časté nežádoucí účinky (mohou postihovat až 1 ze 100 lidí) jsou:
- bolest v horní části břicha, abnormální střevní zvuky, sucho v ústech, nevolnost, nepříjemný pocit v žaludku, těžká zácpa, špatná funkce tenkého střeva (subileus),
- snížená citlivost a vnímavost (hlavně na kůži), smyslové poruchy, potíže s řečí,
- zúžené zorničky, snížená ostrost vidění (ztráta zraku),
- nespavost,
- pomalý tep,
- pískavé zvuky při dýchání, dušnost.
Nežádoucí účinky, jejichž četnost není známa:
- Stevens-Johnsonův syndrom/toxická epidermální nekrolýza (vzácná závažná kožní reakce).
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek EMEND uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Tobolku vyjímejte z blistru až těsně před užitím.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co EMEND obsahuje
- Léčivou látkou je aprepitantum. Jedna tobolka obsahuje aprepitantum 40 mg.
- Pomocnými látkami jsou: sacharóza, mikrokrystalická celulóza (E 460), hyprolóza (E 463), natrium-lauryl-sulfát, želatina, oxid titaničitý (E 171), šelak, hydroxid draselný, černý oxid železitý (E 172) a žlutý oxid železitý (E 172).
Jak EMEND vypadá a co obsahuje toto balení
40mg tvrdá tobolka je neprůhledná s bílým tělem a hořčicově žlutým víčkem; na těle je po obvodu černým inkoustem vytištěno „464“ a „40 mg“.
Tvrdé tobolky EMEND 40 mg se dodávají v následujících velikostech balení:
- Hliníkový blistr obsahující jednu 40mg tobolku
- 5 hliníkových blistrů obsahujících každý jednu 40mg tobolku
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
Výrobce
Merck Sharp & Dohme B. V Waarderweg 39 2031 BN Haarlem Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Eunrapnu
MepK fflapn h floyM Etnrapna EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com
Česká republika
Merck Sharp & Dohme s.r.o.
Tel.: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 53 00 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited
Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Nederland
Merck Sharp & Dohme BV
Tel: 0800 9999000 (+31 23 5153153)
Eesti
Merck Sharp & Dohme OU Tel.: +372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.OB.E.E.
TpA,: +30 210 98 97 300 dpoc_greece@merck.com
Espaňa
Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúnpoq
Merck Sharp & Dohme Cyprus Limited Tp^: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel:+351 21 446 5700 clic@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel:+ 4021 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel:+ 386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy
Puh/Tel: +358 (0) 9 804 650
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Příbalová informace: informace pro uživatele
EMEND 80 mg tvrdé tobolky
aprepitantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje. Jste-li rodičem dítěte užívajícího přípravek EMEND, přečtěte si prosím pečlivě tuto příbalovou informaci.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám nebo Vašemu dítěti. Nedávejte jej žádné další
osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás nebo Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je EMEND a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat nebo podávat
3. Jak se EMEND užívá
4. Možné nežádoucí účinky
5 Jak EMEND uchovávat
6. Obsah balení a další informace
1. Co je EMEND a k čemu se používá
Přípravek EMEND obsahuje léčivou látku aprepitant a patří do skupiny léčiv nazývaných "antagonisté receptoru neurokininu 1 (NK1)". Mozek má zvláštní oblast, která řídí pocit nevolnosti a zvracení. Přípravek EMEND funguje tak, že blokuje signály do této oblasti, čímž omezuje pocit nevolnosti a zvracení.
Tobolky přípravku EMEND se používají u dospělých a dospívajících od 12 let v kombinaci s dalšími léčivy k předcházení nevolnosti a zvracení vyvolanými chemoterapií (léčbou rakoviny), která jsou silnými a středně silnými spouštěči pocitu nevolnosti a zvracení (jako je cisplatina, cyklofosfamid, doxorubicin nebo epirubicin).
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat nebo podávat Neužívejte přípravek EMEND
- jestliže jste Vy nebo Vaše dítě alergický(á)(é) na aprepitant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- spolu s léky obsahujícími pimozid (používá se k léčbě psychiatrických chorob), terfenadin
a astemizol (používají se k léčení senné rýmy a jiných alergických stavů), cisaprid (používá se k léčbě trávicích potíží). Pokud tyto léky Vy nebo Vaše dítě užíváte, informujte o tom lékaře, protože Vaše léčba nebo léčba Vašeho dítěte bude muset být před nasazením přípravku EMEND upravena.
Upozornění a opatření
Před užitím přípravku EMEND nebo podáním tohoto léku dítěti se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Pokud Vy nebo Vaše dítě trpíte onemocněním jater, řekněte to před léčbou tímto přípravkem lékaři, protože játra hrají důležitou roli při rozkladu léku v těle. Proto lékař může potřebovat sledovat stav Vašich jater nebo jater Vašeho dítěte.
Děti a dospívající
Nepodávejte přípravek EMEND 80 mg tobolky dětem mladším 12 let, protože 80mg tobolky nebyly u této populace hodnoceny.
Další léčivé přípravky a přípravek EMEND
Přípravek EMEND může ovlivňovat účinky jiných léků jak během léčby tímto přípravkem, tak po ní. Existují jisté léky, které nesmějí být spolu s přípravkem EMEND užívány (jako je pimozid, terfenadin, astemizol a cisaprid) nebo které vyžadují úpravu dávky (viz také "Neužívejte přípravek EMEND").
Účinky přípravku EMEND nebo jiných léčiv mohou být ovlivněny, pokud budete Vy nebo Vaše dítě přípravek EMEND užívat spolu s dalšími léky, včetně léků uvedených dále. Poraďte se prosím s lékařem nebo lékárníkem, pokud Vy nebo Vaše dítě užíváte některý z následujících léků:
- léky k zabránění početí, které mohou zahrnovat antikoncepční pilulky, kožní náplasti, implantáty a některá nitroděložní tělíska, která uvolňují hormony, nemusí fungovat odpovídajícím způsobem, pokud se používají spolu s přípravkem EMEND. Během léčby přípravkem EMEND a až 2 měsíce po jeho použití je nutno používat jiné nebo dodatečné nehormonální formy antikoncepce,
- cyklosporin, takrolimus, sirolimus, everolimus (imunosupresiva),
- alfentanil, fentanyl (používají se k léčbě bolesti),
- chinidin (používá se k léčbě nepravidelného tepu srdce),
- irinotekan, etoposid, vinorelbin, ifosfamid (léky používané k léčbě rakoviny),
- léky obsahující deriváty ergotových alkaloidů, jako je ergotamin a diergotamin (používané k léčbě migrén),
- warfarin, acenokumarol (léky na ředění krve; může být potřebné provedení krevních testů),
- rifampicin, klarithromycin, telithromycin (antibiotika užívaná k léčbě infekcí),
- fenytoin (lék používaný k léčbě záchvatů),
- karbamazepin (lék používaný k léčbě depresí a epilepsie),
- midazolam, triazolam, fenobarbital (utišující léky nebo léky usnadňující spánek),
- třezalka tečkovaná (bylinný přípravek používaný k léčbě deprese),
- inhibitory proteázy (používají se k léčbě infekcí HIV),
- ketokonazol, kromě šampónu (používá se k léčbě Cushingova syndromu - kdy tělo vytváří nadměrná množství kortizolu)
- itrakonazol, vorikonazol, posakonazol (léky proti plísním),
- nefazodon (používá se k léčbě deprese),
- kortikosteroidy (jako je dexamethason a methylprednisolon),
- léky proti úzkosti (jako je alprazolam),
- tolbutamid (lék používaný k léčbě cukrovky)
Informujte lékaře nebo lékárníka o všech lécích, které Vy nebo Vaše dítě užíváte, které jste v nedávné době užíval(a)(o) nebo které možná budete užívat.
Těhotenství a kojení
Tento přípravek se během těhotenství nesmí užívat, pokud to není nezbytně nutné. Pokud jste Vy nebo Vaše dítě těhotná/těhotné nebo kojíte, domníváte se, že můžete/může být těhotná/těhotné, nebo plánujete/plánuje otěhotnět, poraďte se s lékařem dříve, než začnete/začne tento přípravek užívat.
Ohledně informací týkajících se antikoncepce viz část "Další léčivé přípravky a přípravek EMEND".
Není známo, zda se přípravek EMEND vylučuje do lidského mateřského mléka; proto není doporučeno kojit během léčby tímto přípravkem. Pokud Vy nebo Vaše dítě kojíte/kojí nebo hodláte/hodlá kojit, je třeba sdělit tuto skutečnost lékaři ještě předtím, než začnete/začne užívat tento přípravek.
Řízení dopravních prostředků a obsluha strojů
Je nutno vzít v úvahu, že u některých lidí může po užití přípravku EMEND vzniknout pocit závrati a ospalosti. Pokud Vy nebo Vaše dítě máte/má pocit závrati a ospalosti, nesmíte/nesmí po užití tohoto
přípravku řídit, jezdit na kole, obsluhovat stroje nebo používat nářadí (viz „Možné nežádoucí účinky“).
Přípravek EMEND obsahuje sacharózu
Tobolky přípravku EMEND obsahují sacharózu. Pokud Vám lékař řekl, že Vy nebo Vaše dítě nesnášíte některé cukry, poraďte se s lékařem, než začnete tento přípravek užívat.
3. Jak se EMEND užívá
Vždy užívejte nebo podávejte svému dítěti tento přípravek přesně podle pokynů lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou. Přípravek EMEND vždy užívejte s dalšími léky, aby zabránil pocitu nevolnosti a zvracení. Po léčbě přípravkem EMEND Vás lékař požádá, abyste Vy nebo Vaše dítě pokračoval(a)(o) v užívání dalších léků bránících pocitu nevolnosti a zvracení, včetně kortikosteroidů (jako je dexamethason) a antagonistů „5HT3“ (jako je ondansetron). Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou.
Doporučená perorální dávka (podaná ústy) přípravku EMEND je 1. den:
- jedna 125mg tobolka 1 hodinu před zahájením chemoterapeutického cyklu
a
2. a 3. den:
- jedna 80mg tobolka každý den
- pokud se nepodává žádná chemoterapie, užívejte přípravek EMEND ráno
- pokud se podává chemoterapie, užívejte přípravek EMEND 1 hodinu před zahájením chemoterpeutického cyklu
Přípravek EMEND lze užívat s jídlem nebo bez jídla.
Tobolku spolkněte celou a zapijte ji douškem tekutiny.
Jestliže jste užil(a) více přípravku EMEND, než jste měl(a)
Neberte si více tobolek, než Vám předepsal lékař. Pokud jste nebo Vaše dítě užil(a)(o) více tobolek, okamžitě vyhledejte svého lékaře.
Jestliže jste zapomněl(a) užít EMEND
Pokud Vy nebo Vaše dítě vynecháte dávku, poraďte se se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přestaňte přípravek EMEND užívat a ihned navštivte lékaře, pokud Vy nebo Vaše dítě zaznamenáte kterýkoli z následujících nežádoucích účinků, které mohou být závažné a kvůli nimž můžete Vy nebo Vaše dítě naléhavě potřebovat lékařské ošetření:
- kopřivka, vyrážka, svědění, potíže s dýcháním nebo polykáním (četnost není známa, z dostupných údajů ji nelze určit); jde o známky alergické reakce.
Další hlášené nežádoucí účinky jsou uvedeny dále.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 lidí) jsou:
- zácpa, poruchy trávení,
- bolest hlavy,
- únava,
- ztráta chuti k jídlu,
- škytavka,
- zvýšené množství jaterních enzymů v krvi.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 lidí) jsou:
- závrať, ospalost,
- akné, vyrážka,
- úzkost,
- říhání, pocit nevolnosti, zvracení, pálení žáhy, bolest žaludku, sucho v ústech, plynatost,
- častější, bolestivé nebo pálivé močení,
- slabost, celkový pocit nepohody,
- návaly horka/zarudnutí obličeje nebo kůže,
- rychlý nebo nepravidelný tep,
- horečka se zvýšeným rizikem infekce, snížení počtu červených krvinek.
Vzácné nežádoucí účinky (mohou postihnout až 1 z 1 000 lidí) jsou:
- potíže s přemýšlením, nedostatek energie, poruchy vnímání chutí,
- citlivost kůže na sluneční světlo, nadměrné pocení, mastná kůže, boláky na kůži, svědivá vyrážka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza (vzácná, závažná kožní reakce),
- euforie (pocit nadměrného štěstí), dezorientace,
- bakteriální infekce, plísňové infekce,
- těžká zácpa, žaludeční vředy, zánět tenkého střeva a tlustého střeva, boláky v ústech, nadýmání,
- časté močení, větší výdej moči, než je obvyklé, přítomnost cukru nebo krve v moči,
- pocit nepohody na hrudi, otok, změna způsobu chůze,
- kašel, hlen v zadní části hrdla (tzv. zadní rýma), podráždění hrdla, kýchání, bolest v krku,
- výtok z očí a svědění očí,
- zvonění v uších,
- svalové křeče, svalová slabost,
- nadměrná žízeň,
- pomalý srdeční tep, onemocnění srdce a cév,
- pokles počtu bílých krvinek, nízké hladiny sodíku v krvi, pokles tělesné hmotnosti.
Hlášení nežádoucích účinků
Pokud se u Vás nebo Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek EMEND uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Tobolku vyjímejte z blistru až těsně před užitím.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co EMEND obsahuje
- Léčivou látkou je aprepitantum. Jedna tobolka obsahuje aprepitantum 80 mg.
- Pomocnými látkami jsou: sacharóza, mikrokrystalická celulóza (E 460), hyprolóza (E 463), natrium-lauryl-sulfát, želatina, oxid titaničitý (E 171), šelak, hydroxid draselný a černý oxid železitý (E 172).
Jak EMEND vypadá a co obsahuje toto balení
80mg tvrdá tobolka je neprůhledná s víčkem a tělem bílé barvy; na těle je po obvodu černým inkoustem vytištěno „461“ a „80 mg“.
Tvrdé tobolky EMEND 80 mg se dodávají v následujících velikostech balení:
• hliníkový blistr obsahující jednu 80mg tobolku
• balení pro 2-denní léčbu obsahující dvě 80mg tobolky
• 5 hliníkových blistrů obsahujících každý jednu 80mg tobolku
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
Výrobce
Merck Sharp & Dohme B. V Waarderweg 39 2031 BN Haarlem Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Eunrapnu
MepK fflapn h floyM Etnrapnn EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com
Česká republika
Merck Sharp & Dohme s.r.o.
Tel.: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 53 00 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited
Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OU Tel.:+372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.OB.E.E.
TpA,: +30 210 98 97 300 dpoc_greece@merck.com
Espaňa
Merck Sharp & Dohme de Espana, S.A.
Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúnpoq
Merck Sharp & Dohme Cyprus Limited Tp^: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Nederland
Merck Sharp & Dohme BV
Tel: 0800 9999000 (+31 23 5153153)
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel:+351 21 446 5700 clic@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel:+ 4021 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel:+ 386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy
Puh/Tel: +358 (0) 9 804 650
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Příbalová informace: informace pro uživatele
EMEND 125 mg tvrdé tobolky
aprepitantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje. Jste-li rodičem dítěte užívajícího přípravek EMEND, přečtěte si prosím pečlivě tuto příbalovou informaci.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám nebo Vašemu dítěti. Nedávejte jej žádné další
osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás nebo Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je EMEND a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat nebo podávat
3. Jak se EMEND užívá
4. Možné nežádoucí účinky
5 Jak EMEND uchovávat
6. Obsah balení a další informace
1. Co je EMEND a k čemu se používá
Přípravek EMEND obsahuje léčivou látku aprepitant a patří do skupiny léčiv nazývaných "antagonisté receptoru neurokininu 1 (NK1)". Mozek má zvláštní oblast, která řídí pocit nevolnosti a zvracení. Přípravek EMEND funguje tak, že blokuje signály do této oblasti, čímž omezuje pocit nevolnosti a zvracení.
Tobolky přípravku EMEND se používají u dospělých a dospívajících od 12 let věku v kombinaci s dalšími léčivy k předcházení nevolnosti a zvracení vyvolanými chemoterapií (léčbou rakoviny), která je silným a středně silným spouštěčem pocitu nevolnosti a zvracení (jako je cisplatina, cyklofosfamid, doxorubicin nebo epirubicin).
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat nebo podávat Neužívejte přípravek EMEND
- jestliže jste Vy nebo Vaše dítě alergický(á)(é) na aprepitant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- spolu s léky obsahujícími pimozid (používá se k léčení psychiatrických chorob), terfenadin
a astemizol (používají se k léčení senné rýmy a jiných alergických stavů), cisaprid (používá se k léčbě trávicích potíží). Pokud tyto léky Vy nebo Vaše dítě užíváte, informujte o tom lékaře, protože Vaše léčba nebo léčba Vašeho dítěte bude muset být před nasazením přípravku EMEND upravena.
Upozornění a opatření
Před užitím přípravku EMEND nebo podáním tohoto léku dítěti se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Pokud Vy nebo Vaše dítě trpíte onemocněním jater, řekněte to před léčbou přípravkem EMEND lékaři, protože játra hrají důležitou roli při rozkladu léku v těle. Proto lékař může potřebovat sledovat stav Vašich jater nebo jater Vašeho dítěte.
Děti a dospívající
Nepodávejte přípravek EMEND 125 mg tobolky dětem mladším 12 let, protože 125mg tobolky nebyly u této populace hodnoceny.
Další léčivé přípravky a přípravek EMEND
Přípravek EMEND může ovlivňovat účinky jiných léků jak během léčby tímto přípravkem, tak po ní. Existují jisté léky, které nesmějí být spolu s přípravkem EMEND užívány (jako je pimozid, terfenadin, astemizol a cisaprid) nebo které vyžadují úpravu dávky (viz také "Neužívejte přípravek EMEND").
Účinky přípravku EMEND nebo jiných léčiv mohou být ovlivněny, pokud budete Vy nebo Vaše dítě přípravek EMEND užívat spolu s dalšími léky, včetně léků uvedených dále. Poraďte se prosím s lékařem nebo lékárníkem, pokud Vy nebo Vaše dítě užíváte některý z následujících léků:
- léky k zabránění početí, které mohou zahrnovat antikoncepční pilulky, kožní náplasti, implantáty a některá nitroděložní tělíska, která uvolňují hormony, nemusí fungovat odpovídajícím způsobem, pokud se používají spolu s přípravkem EMEND. Během léčby přípravkem EMEND a až 2 měsíce po jeho použití je nutno používat jiné nebo dodatečné nehormonální formy antikoncepce,
- cyklosporin, takrolimus, sirolimus, everolimus (imunosupresiva),
- alfentanil, fentanyl (používají se k léčbě bolesti),
- chinidin (používá se k léčbě nepravidelného tepu srdce),
- irinotekan, etoposid, vinorelbin, ifosfamid (léky používané k léčbě rakoviny),
- léky obsahující deriváty ergotových alkaloidů, jako je ergotamin a diergotamin (používané k léčbě migrén),
- warfarin, acenokumarol (léky na ředění krve; může být potřebné provedení krevních testů),
- rifampicin, klarithromycin, telithromycin (antibiotika užívaná k léčbě infekcí),
- fenytoin (lék používaný k léčbě záchvatů),
- karbamazepin (lék používaný k léčbě depresí a epilepsie),
- midazolam, triazolam, fenobarbital (utišující léky nebo léky usnadňující spánek),
- třezalka tečkovaná (bylinný přípravek používaný k léčbě deprese),
- inhibitory proteázy (používají se k léčbě infekcí HIV),
- ketokonazol, kromě šampónu (používá se k léčbě Cushingova syndromu - kdy tělo vytváří nadměrná množství kortizolu)
- itrakonazol, vorikonazol, posakonazol (léky proti plísním),
- nefazodon (používá se k léčbě deprese),
- kortikosteroidy (jako je dexamethason a methylprednisolon),
- léky proti úzkosti (jako je alprazolam),
- tolbutamid (lék používaný k léčbě cukrovky)
Informujte lékaře nebo lékárníka o všech lécích, které Vy nebo Vaše dítě užíváte, které jste v nedávné době užíval(a)(o) nebo které možná budete užívat.
Těhotenství a kojení
Tento přípravek se během těhotenství nesmí užívat, pokud to není nezbytně nutné. Pokud jste Vy nebo Vaše dítě těhotná/těhotné nebo kojíte, domníváte se, že můžete/může být těhotná/těhotné, nebo plánujete/plánuje otěhotnět, poraďte se s lékařem dříve, než začnete/začne tento přípravek užívat.
Ohledně informací týkajících se antikoncepce viz část "Další léčivé přípravky a přípravek EMEND".
Není známo, zda se EMEND vylučuje do lidského mateřského mléka; proto není doporučeno kojit během léčby tímto přípravkem. Pokud kojíte nebo hodláte kojit, je třeba sdělit tuto skutečnost lékaři ještě předtím, než začnete užívat tento přípravek.
Řízení dopravních prostředků a obsluha strojů
Je nutno vzít v úvahu, že u některých lidí může po užití přípravku EMEND vzniknout pocit závrati a ospalosti. Pokud Vy nebo Vaše dítě máte pocit závrati a ospalosti, nesmíte po užití tohoto přípravku řídit, jezdit na kole, obsluhovat stroje ani používat nářadí (viz „Možné nežádoucí účinky“).
Příp ravek EMEND obsahuje sacharózu
Tobolky přípravku EMEND obsahují sacharózu. Pokud Vám lékař řekl, že Vy nebo Vaše dítě nesnášíte některé cukry, poraďte se s lékařem, než začnete tento přípravek užívat.
3. Jak se EMEND užívá
Vždy užívejte nebo podávejte svému dítěti tento přípravek přesně podle pokynů lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou. Přípravek EMEND vždy užívejte s dalšími léky, aby zabránil pocitu nevolnosti a zvracení. Po léčbě přípravkem EMEND Vás lékař požádá, abyste Vy nebo Vaše dítě pokračoval(a)(o) v užívání dalších léků bránících pocitu nevolnosti a zvracení, včetně kortikosteroidů (jako je dexamethason) a antagonistů „5HT3“ (jako je ondansetron). Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou.
Doporučená perorální dávka (podaná ústy) přípravku EMEND je
1. den:
- jedna 125mg tobolka 1 hodinu před zahájením chemoterapeutického cyklu a
2. a 3. den:
- jedna 80mg tobolka každý den
- pokud se nepodává žádná chemoterapie, užívejte přípravek EMEND ráno
- pokud se podává chemoterapie, užívejte přípravek EMEND 1 hodinu před zahájením chemoterpeutického cyklu
Přípravek EMEND lze užívat s jídlem nebo bez jídla.
Tobolku spolkněte celou a zapijte ji douškem tekutiny.
Jestliže jste užil(a) více přípravku EMEND, než jste měl(a)
Neberte si více tobolek, než Vám předepsal lékař. Pokud byste si Vy nebo Vaše dítě vzal(a)(o)i více tobolek, okamžitě vyhledejte svého lékaře.
Jestliže jste zapomněl(a) užít EMEND
Pokud Vy nebo Vaše dítě vynecháte dávku, poraďte se se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přestaňte přípravek EMEND užívat a ihned navštivte lékaře, pokud Vy nebo Vaše dítě zaznamenáte kterýkoli z následujících nežádoucích účinků, které mohou být závažné a kvůli nimž můžete Vy nebo Vaše dítě naléhavě potřebovat lékařské ošetření:
- kopřivka, vyrážka, svědění, potíže s dýcháním nebo polykáním (četnost není známa, z dostupných údajů ji nelze určit); jde o známky alergické reakce.
Další hlášené nežádoucí účinky jsou uvedeny dále.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 lidí) jsou:
- zácpa, poruchy trávení,
- bolest hlavy,
- únava,
- ztráta chuti k jídlu,
- škytavka,
- zvýšené množství jaterních enzymů v krvi.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 lidí) jsou:
- závrať, ospalost,
- akné, vyrážka,
- úzkost,
- říhání, pocit nevolnosti, zvracení, pálení žáhy, bolest žaludku, sucho v ústech, plynatost,
- častější, bolestivé nebo pálivé močení,
- slabost, celkový pocit nepohody,
- návaly horka/zarudnutí obličeje nebo kůže,
- rychlý nebo nepravidelný tep,
- horečka se zvýšeným rizikem infekce, snížení počtu červených krvinek.
Vzácné nežádoucí účinky (mohou postihnout až 1 z 1 000 lidí) jsou:
- potíže s přemýšlením, nedostatek energie, poruchy vnímání chutí,
- citlivost kůže na sluneční světlo, nadměrné pocení, mastná kůže, boláky na kůži, svědivá vyrážka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza (vzácná, závažná kožní reakce),
- euforie (pocit nadměrného štěstí), dezorientace,
- bakteriální infekce, plísňové infekce,
- těžká zácpa, žaludeční vředy, zánět tenkého střeva a tlustého střeva, boláky v ústech, nadýmání,
- časté močení, větší výdej moči, než je obvyklé, přítomnost cukru nebo krve v moči,
- pocit nepohody na hrudi, otok, změna způsobu chůze,
- kašel, hlen v zadní části hrdla (tzv. zadní rýma), podráždění hrdla, kýchání, bolest v krku,
- výtok z očí a svědění očí,
- zvonění v uších,
- svalové křeče, svalová slabost,
- nadměrná žízeň,
- pomalý srdeční tep, onemocnění srdce a cév,
- pokles počtu bílých krvinek, nízké hladiny sodíku v krvi, pokles tělesné hmotnosti.
Hlášení nežádoucích účinků
Pokud se u Vás nebo Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek EMEND uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Tobolku vyjímejte z blistru až těsně před užitím.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co EMEND obsahuje
- Léčivou látkou je aprepitantum. Jedna 125mg tvrdá tobolka obsahuje aprepitantum 125 mg.
- Pomocnými látkami jsou: sacharóza, mikrokrystalická celulóza (E 460), hyprolóza (E 463), natrium-lauryl-sulfát, želatina, oxid titaničitý (E 171), šelak, hydroxid draselný, černý oxid železitý (E 172), červený oxid železitý (E 172) a žlutý oxid železitý (E 172).
Jak EMEND vypadá a co obsahuje toto balení
125mg tvrdá tobolka je neprůhledná s bílým tělem a růžovým víčkem; na těle je po obvodu černým inkoustem vytištěno „462“ a „125 mg“.
Tvrdé tobolky EMEND 125 mg se dodávají v následujících velikostech balení:
- hliníkový blistr obsahující jednu 125mg tobolku
- 5 hliníkových blistrů obsahujících každý jednu 125mg tobolku
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
Výrobce
Merck Sharp & Dohme B. V Waarderweg 39 2031 BN Haarlem Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Eunrapnu
MepK fflapn h floyM Etnrapna EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com
Česká republika
Merck Sharp & Dohme s.r.o.
Tel.: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 53 00 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited
Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Nederland
Merck Sharp & Dohme BV
Tel: 0800 9999000 (+31 23 5153153)
Eesti
Merck Sharp & Dohme OU Tel.:+372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.OB.E.E.
TpA,: +30 210 98 97 300 dpoc_greece@merck.com
Espaňa
Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúnpoq
Merck Sharp & Dohme Cyprus Limited Tp^: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel:+351 21 446 5700 clic@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel:+ 4021 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel:+ 386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy
Puh/Tel: +358 (0) 9 804 650
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Příbalová informace: informace pro uživatele
EMEND 165 mg tvrdé tobolky
aprepitantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je přípravek EMEND a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek EMEND užívat
3. Jak se přípravek EMEND užívá
4. Možné nežádoucí účinky
5 Jak přípravek EMEND uchovávat
6. Obsah balení a další informace
1. Co je přípravek EMEND a k čemu se používá
Přípravek EMEND obsahuje léčivou látku aprepitant, která patří do skupiny léků nazývaných "antagonisté receptoru neurokininu 1 (NKi)". Mozek má specifickou oblast, která ovládá pocit nevolnosti a zvracení. Přípravek EMEND působí prostřednictvím blokády signálů do této oblasti, čímž snižuje pocit nevolnosti a zvracení. EMEND se používá u dospělých v kombinaci s dalšími léčivy k předcházení nevolnosti a zvracení vyvolanými chemoterapií (léčbou rakoviny) obsahující cisplatinu (silný spouštěč pocitu nevolnosti a zvracení) a s chemoterapií, které jsou středně silným spouštěčem pocitu nevolnosti a zvracení (jako je cyklofosfamid, doxorubicin nebo epirubicin).
2. Čemu musíte věnovat pozornost, než začnete přípravek EMEND užívat Neužívejte přípravek EMEND
- jestliže jste alergický(á) na aprepitant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- spolu s léky obsahujícími pimozid (používá se k léčbě psychiatrických nemocí), terfenadin a astemizol (používá se k léčbě senné rýmy a jiných alergických stavů), cisaprid (používá se k léčbě trávicích potíží). Pokud tyto léky užíváte, informujte o tom svého ošetřujího lékaře, protože Vaše léčba musí být předtím, než začnete používat přípravek EMEND upravena.
Upozornění a opatření
Před užitím tohoto přípravku se poraďte se svým lékařem, lékarníkem nebo zdravotní sestrou.
Pokud máte onemocnění jater, sdělte to před léčbou přípravkem EMEND svému lékaři, protože Vaše játra hrají důležitou roli při rozkládání léčiva v těle. Proto může Váš lékař považovat za nezbytné sledovat stav Vašich jater.
Děti a dospívající
Nepodávejte přípravek EMEND 165 mg dětem a dospívajícím mladším 18 let, protože 165mg tobolky nebyly u této populace hodnoceny.
Další léčivé přípravky a přípravek EMEND
Přípravek EMEND může ovlivnit jiné léky, a to jak během léčby nebo po léčbě přípravkem EMEND. Existují určité léky, které se nesmí užívat spolu s přípravkem EMEND (jako je pimozid, terfenadin, astemizol a cisaprid) nebo které vyžadují úpravu dávky (viz také Nepoužívejte přípravek EMEND).
Účinky přípravku EMEND nebo jiných léčiv mohou být ovlivněny, pokud budete přípravek EMEND užívat spolu s dalšími léky, včetně léků uvedených dále. Poraďte se prosím se svým lékařem nebo lékárníkem, pokud užíváte některý z následujících léků:
- léky k zabránění početí, které mohou zahrnovat antikoncepční pilulky, kožní náplasti, implantáty a některá nitroděložní tělíska (IUD), která uvolňují hormony, nemusí fungovat odpovídajícím způsobem, pokud se používají spolu s přípravkem EMEND. Během léčby přípravkem EMEND a až 2 měsíce po jeho použití je nutno používat jiné nebo dodatečné nehormonální formy antikoncepce,.
- cyklosporin, takrolimus, sirolimus, everolimus (imunosupresiva),
- alfentanil, fentanyl (používají se k léčbě bolesti),
- chinidin (používá se k léčbě nepravidelného tepu srdce),
- irinotekan, etoposid, vinorelbin, ifosfamid (léky používané k léčbě rakoviny),
- léky obsahující deriváty námelových alkaloidů, jako je ergotamin a diergotamin (používané k léčbě migrén),
- warfarin, acenokumarol (léky na ředění krve; může být požadováno provedení krevních testů),
- rifampicin, klarithromycin, telithromycin (antibiotika užívaná k léčbě infekcí),
- fenytoin (léčivý přípravek používaný k léčbě záchvatů),
- karbamazepin (léčivý přípravek používaný k léčbě depresí a epilepsie),
- midazolam, triazolam, fenobarbital (utišující léčivé přípravky nebo léčivé přípravky usnadňující spánek),
- třezalka tečkovaná (bylinný přípravek používaný k léčbě deprese),
- inhibitory proteázy (používají se k léčbě infekcí HIV),
- ketokonazol, kromě šampónu (používá se k léčbě Cushingova syndromu - kdy tělo vytváří nadměrná množství kortizolu)
- itrakonazol, vorikonazol, posakonazol (léky proti plísním),
- nefazodon (používá se k léčbě deprese),
- kortikosteroidy (jako je dexamethason a methylprednisolon),
- léky proti úzkosti (jako je alprazolam),
- tolbutamid (léčivý přípravek používaný k léčbě cukrovky)
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Těhotenství a kojení
Tento přípravek během těhotenství nesmíte užívat, pokud to není nezbytně nutné. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek užívat.
Informace ohledně antikoncepce viz část „Další léčivé přípravky a přípravek EMEND“
Není známo, zda se přípravek EMEND vylučuje do lidského mateřského mléka a tudíž se během léčby tímto přípravkem kojení nedoporučuje. Je důležité, abyste před použitím tohoto přípravku svého ošetřujícího lékaře informovala, že kojíte nebo že kojení plánujete.
Řízení dopravních prostředků a obsluha strojů
Je nutno vzít v úvahu, že někteří lidé po použití přípravku EMEND mají závratě nebo cítí ospalost. Jestliže máte závratě nebo cítíte ospalost, nesmíte po užití tohoto přípravku řídit nebo obsluhovat stroje (viz „Možné nežádoucí účinky“).
Přípravek EMEND obsahuje sacharózu
Tobolky přípravku EMEND obsahují sacharózu. Pokud Vám Váš lékař řekl, že nesnášíte některé cukry, poraďte se se svým lékařem , než začnete tento léčivý přípravek užívat.
3. Jak se přípravek EMEND užívá
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře nebo lékárníka. Přípravek EMEND vždy užívejte s dalšími léky, aby zabránil pocitu nevolnosti a zvracení. Po léčbě přípravkem EMEND Vás lékař požádá, abyste následující tři dny pokračoval(a) v užívání dalších léků bránících pocitu nevolnosti a zvracení, včetně kortikosteroidů (jako je dexamethason) a antagonistů „5HT3“ (jako je ondansetron).
Doporučená dávka přípravku EMEND je jedna 165mg tobolka první den cyklu chemoterapeutické léčby. Užijte 1 tobolku 1 hodinu před zahájením chemoterapeutického cyklu.
Přípravek EMEND lze užívat s jídlem i bez jídla.
Tobolku polkněte celou a zapijte ji trochou tekutiny.
Jestliže jste užil(a) více přípravku EMEND, než jste měl(a)
Neužívejte více tobolek, než Vám doporučil lékař. Pokud jste užil/a příliš mnoho tobolek, ihned se obraťte na svého lékaře.
Jestliže jste zapomněl(a) užít přípravek EMEND
Jestliže jste vynechal/a dávku, poraďte se se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto léku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přestaňte užívat přípravek EMEND a ihned navštivte lékaře, pokud zaznamenáte kterýkoli z následujících nežádoucích účinků, jež mohou být závažné a kvůli nimž můžete potřebovat naléhavé lékařské ošetření:
- kopřivka, vyrážka, svědění, potíže s dýcháním nebo polykáním - (četnost není známa, z dostupných údajů ji nelze odhadnout) jde o příznaky alergické reakce.
Další hlášené nežádoucí účinky jsou uvedeny dále.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 lidí) jsou:
- zácpa, poruchy trávení,
- bolest hlavy,
- únava,
- ztráta chuti k jídlu,
- škytavka,
- zvýšené hodnot jaterních testů.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 lidí) jsou:
- závrať, ospalost,
- akné, vyrážka,
- úzkost,
- říhání, pocit nevolnosti, zvracení, pálení žáhy, bolest břicha, sucho v ústech, plynatost,
- častější, bolestivé nebo pálivé močení
- celkový pocit nemoci
- návaly horka
- rychlý nebo nepravidelný srdeční tep
- horečka se zvýšeným rizikem infekce, snížení počtu červených krvinek.
Vzácné nežádoucí účinky (mohou postihnout až 1 z 1 000 lidí) jsou:
- potíže s přemýšlením, nedostatek energie, poruchy vnímání chutí,
- citlivost kůže na sluneční světlo, nadměrné pocení, mastná kůže, boláky na kůži, svědivá vyrážka, Stevens—Johnsonův syndrom/toxická epidermální nekrolýza (vzácná, závažná kožní reakce),
- euforie (pocit nadměrného štěstí), dezorientace,
- bakteriální infekce, plísňové infekce,
- těžká zácpa, žaludeční vředy, zánět tenkého střeva a tlustého střeva, boláky v ústech, nadýmání,
- časté močení, větší výdej moči, než je obvyklé, přítomnost cukru nebo krve v moči,
- nepříjemný pocit na hrudi, otok, změna způsobu chůze,
- kašel, hlen v zadní části hrdla (tzv. zadní rýma), podráždění hrdla, kýchání, bolest v krku,
- výtok z očí a svědění očí,
- zvonění v uších,
- svalové křeče, svalová slabost,
- nadměrná žízeň,
- pomalý srdeční tep, onemocnění srdce a cév,pokles počtu bílých krvinek, nízké hladiny sodíku v krvi, pokles tělesné hmotnosti.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékař, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek EMEND uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na blistru a krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu aby byl přípravek chráněn před vlhkostí.
Tobolku z blistru nevyjímejte dokud nebudete připraven(a) ji užít.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek EMEND obsahuje
- Léčivou látkou je aprepitantum. Jedna tobolka obsahuje aprepitantum 165 mg.
- Pomocnými látkami jsou: sacharóza, mikrokrystalická celulóza (E 460), hyprolóza (E 463), natrium-lauryl-sulfát, želatina, oxid titaničitý (E 171), indigokarmín (E 132), šelak, hydroxid draselný, černý oxid železitý (E 172).
Jak přípravek EMEND vypadá a co obsahuje toto balení
165mg tvrdá tobolka je neprůhledná se světle modrým víčkem a bílým tělem s „466“ a „165 mg“ vytištěným radiálně černým inkoustem na jedné straně těla a logem Merck na druhé.
Tvrdé tobolky EMEND 165 mg se dodávají v papírových krabičkách v následujících velikostech balení:
- hliníkový blistr obsahující jednu 165mg tobolku
- 6 hliníkových blistrů, každý obsahuje jednu 165mg tobolku
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
|
Držitel rozhodnutí o registraci Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN11 9BU Velká Británie |
Výrobce Merck Sharp & Dohme B. V. Waarderweg 39 2031 BN Haarlem Nizozemsko |
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgique/Belgie/Belgien MSD Belgium BVBA/SPRL Tél/Tel: 0800 38 693 (+32(0)27766211) |
Lietuva UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com |
|
Eunrapnu MepK fflapn h floyM Eturapna EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com |
Luxembourg/Luxemburg MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com |
|
Česká republika Merck Sharp & Dohme s.r.o. Tel.: +420 233 010 111 dpoc_czechslovak@merck.com |
Magyarország MSD Pharma Hungary Kft. Tel.: +36 1 888 53 00 hungary_msd@merck.com |
|
Danmark MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com |
Malta Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com |
|
Deutschland MSD SHARP & DOHME GMBH Tel: 0800 673 673 673 (+49 (0) 89 4561 2612) |
Nederland Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) |
|
Eesti Merck Sharp & Dohme OU Tel.:+372 6144 200 msdeesti@merck.com |
Norge MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no |
EXXáSa
MSD A.OB.E.E.
TpA,: +30 210 98 97 300 dpoc_greece@merck.com
Espaňa
Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúnpoq
Merck Sharp & Dohme Cyprus Limited Tp^: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp. z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel:+351 21 446 5700 clic@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel:+ 4021 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel:+ 386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy
Puh/Tel: +358 (0) 9 804 650
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Příbalová informace: informace pro uživatele
EMEND 125 mg tvrdé tobolky EMEND 80 mg tvrdé tobolky
aprepitantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje. Jste-li rodičem dítěte užívajícího přípravek EMEND, přečtěte si prosím pečlivě tuto příbalovou informaci.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vám nebo Vašemu dítěti. Nedávejte jej žádné další
osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás nebo Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci:
1. Co je EMEND a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat nebo podávat
3. Jak se EMEND užívá
4. Možné nežádoucí účinky
5 Jak EMEND uchovávat
6. Obsah balení a další informace
1. Co je EMEND a k čemu se používá
Přípravek EMEND obsahuje léčivou látku aprepitant a patří do skupiny léčiv nazývaných "antagonisté receptoru neurokininu 1 (NK1)". Mozek má zvláštní oblast, která řídí pocit nevolnosti a zvracení. Přípravek EMEND funguje tak, že blokuje signály do této oblasti, čímž omezuje pocit nevolnosti a zvracení.
Tobolky přípravku EMEND se používají u dospělých a dospívajících od 12 let věku v kombinaci s dalšími léčivy k předcházení nevolnosti a zvracení vyvolanými chemoterapií (léčbou rakoviny), která je silným a středně silným spouštěčem pocitu nevolnosti a zvracení (jako je cisplatina, cyklofosfamid, doxorubicin nebo epirubicin).
2. Čemu musíte věnovat pozornost, než začnete EMEND užívat nebo podávat Neužívejte přípravek EMEND
- jestliže jste Vy nebo Vaše dítě alergický(á)(é) na aprepitant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- spolu s léky obsahujícími pimozid (používá se k léčbě psychiatrických chorob), terfenadin
a astemizol (používají se k léčení senné rýmy a jiných alergických stavů), cisaprid (používá se k léčbě trávicích potíží). Pokud tyto léky Vy nebo Vaše dítě užíváte, informujte o tom lékaře, protože Vaše léčba nebo léčba Vašeho dítěte bude muset být před nasazením přípravku EMEND upravena.
Upozornění a opatření
Před užitím přípravku EMEND nebo podáním tohoto přípravku dítěti se poraďte se svým lékařem, lékárníkem nebo zdravotní sestrou.
Pokud Vy nebo Vaše dítě trpíte onemocněním jater, řekněte to před léčbou přípravkem EMEND lékaři, protože játra hrají důležitou roli při rozkladu léku v těle. Proto lékař může potřebovat sledovat stav Vašich jater nebo jater Vašeho dítěte.
Děti a dospívající
Nepodávejte přípravek EMEND 80 mg a 125 mg dětem a dospívajícím mladším 18 let, protože 165mg tobolky nebyly u této populace hodnoceny.
Další léčivé přípravky a přípravek EMEND
Přípravek EMEND může ovlivňovat účinkyjiných léků jak během léčby tímto přípravkem, tak po ní. Existují jisté léky, které nesmějí být spolu s přípravkem EMEND užívány (jako je pimozid, terfenadin, astemizol a cisaprid) nebo které vyžadují úpravu dávky (viz také "Neužívejte přípravek EMEND").
Účinky přípravku EMEND nebo jiných léčiv mohou být ovlivněny, pokud budete Vy nebo Vaše dítě přípravek EMEND užívat spolu s dalšími léky, včetně léků uvedených dále. Poraďte se prosím s lékařem nebo lékárníkem, pokud Vy nebo Vaše dítě užíváte některý z následujících léků:
- léky k zabránění početí, které mohou zahrnovat antikoncepční pilulky, kožní náplasti, implantáty a některá nitroděložní tělíska, která uvolňují hormony, nemusí fungovat odpovídajícím způsobem, pokud se používají spolu s přípravkem EMEND. Během léčby přípravkem EMEND a až 2 měsíce po jeho použití je nutno používat jiné nebo dodatečné nehormonální formy antikoncepce,
- cyklosporin, takrolimus, sirolimus, everolimus (imunosupresiva),
- alfentanil, fentanyl (používají se k léčbě bolesti),
- chinidin (používá se k léčbě nepravidelného tepu srdce),
- irinotekan, etoposid, vinorelbin, ifosfamid (léky používané k léčbě rakoviny),
- léky obsahující deriváty ergotových alkaloidů, jako je ergotamin a diergotamin (používané k léčbě migrén),
- warfarin, acenokumarol (léky na ředění krve; může být potřebné provedení krevních testů),
- rifampicin, klarithromycin, telithromycin (antibiotika užívaná k léčbě infekcí),
- fenytoin (lék používaný k léčbě záchvatů),
- karbamazepin (lék používaný k léčbě depresí a epilepsie),
- midazolam, triazolam, fenobarbital (utišující léky nebo léky usnadňující spánek),
- třezalka tečkovaná (bylinný přípravek používaný k léčbě deprese),
- inhibitory proteázy (používají se k léčbě infekcí HIV),
- ketokonazol, kromě šampónu (používá se k léčbě Cushingova syndromu - kdy tělo vytváří nadměrná množství kortizolu)
- itrakonazol, vorikonazol, posakonazol (léky proti plísním),
- nefazodon (používá se k léčbě deprese),
- kortikosteroidy (jako je dexamethason a methylprednisolon),
- léky proti úzkosti (jako je alprazolam),
- tolbutamid (lék používaný k léčbě cukrovky)
Informujte lékaře nebo lékárníka o všech lécích, které Vy nebo Vaše dítě užíváte, které jste v nedávné době užíval(a)(o) nebo které možná budete užívat.
Těhotenství a kojení
Tento přípravek se během těhotenství nesmí užívat, pokud to není nezbytně nutné. Pokud jste Vy nebo Vaše dítě těhotná/těhotné nebo kojíte, domníváte se, že můžete/může být těhotná/těhotné, nebo plánujete/plánuje otěhotnět, poraďte se s lékařem dříve, než začnete/začne tento přípravek užívat.
Ohledně informací týkajících se antikoncepce viz část „Další léčivé přípravky a přípravek EMEND“.
Není známo, zda se EMEND vylučuje do lidského mateřského mléka; proto není doporučeno kojit během léčby tímto přípravkem. Pokud kojíte nebo hodláte kojit, je třeba sdělit tuto skutečnost lékaři ještě předtím, než začnete užívat tento přípravek.
Řízení dopravních prostředků a obsluha strojů
Je nutno vzít v úvahu, že u některých lidí může po užití přípravku EMEND vzniknout pocit závrati a ospalosti. Pokud Vy nebo Vaše dítě máte pocit závrati a ospalosti, nesmíte po užití tohoto přípravku řídit, jezdit na kole, obsluhovat stroje ani používat nářadí (viz „Možné nežádoucí účinky“).
Přípravek EMEND obsahuje sacharózu
Tobolky přípravku EMEND obsahují sacharózu. Pokud Vám lékař řekl , že Vy nebo Vaše dítě nesnášíte některé cukry, poraďte se s lékařem, než začnete tento přípravek užívat.
3. Jak se EMEND užívá
Vždy užívejte nebo podávejte svému dítěti tento přípravek přesně podle pokynů lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou. Přípravek EMEND vždy užívejte s dalšími léky, aby zabránil pocitu nevolnosti a zvracení. Po léčbě přípravkem EMEND Vás lékař požádá, abyste Vy nebo Vaše dítě pokračoval(a)(o) v užívání dalších léků bránících pocitu nevolnosti a zvracení, včetně kortikosteroidů (jako je dexamethason) a antagonistů „5HT3“ (jako je ondansetron). Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou.
Doporučená perorální dávka (podaná ústy) přípravku EMEND je 1. den:
- jedna 125mg tobolka 1 hodinu před zahájením chemoterapeutického cyklu
a
2. a 3. den:
- jedna 80mg tobolka každý den
- pokud se nepodává žádná chemoterapie, užívejte přípravek EMEND ráno
- pokud se podává chemoterapie, užívejte přípravek EMEND 1 hodinu před zahájením chemoterapeutického cyklu
Přípravek EMEND lze užívat s jídlem nebo bez jídla.
Tobolku spolkněte celou a zapijte ji douškem tekutiny.
Jestliže jste užil(a) více přípravku EMEND, než jste měl(a)
Neberte si více tobolek, než Vám předepsal lékař. Pokud byste si Vy nebo Vaše dítě vzal(a)(o)i více tobolek, okamžitě vyhledejte svého lékaře.
Jestliže jste zapomněl(a) užít EMEND
Pokud Vy nebo Vaše dítě vynecháte dávku, poraďte se se svým lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Přestaňte přípravek EMEND užívat a ihned navštivte lékaře, pokud Vy nebo Vaše dítě zaznamenáte kterýkoli z následujících nežádoucích účinků, které mohou být závažné a kvůli nimž můžete Vy nebo Vaše dítě naléhavě potřebovat lékařské ošetření:
- kopřivka, vyrážka, svědění, potíže s dýcháním nebo polykáním (četnost není známa, z dostupných údajů ji nelze určit); jde o známky alergické reakce.
Další hlášené nežádoucí účinky jsou uvedeny dále.
Časté nežádoucí účinky (mohou postihnout až 1 z 10 lidí) jsou:
- zácpa, poruchy trávení,
- bolest hlavy,
- únava,
- ztráta chuti k jídlu,
- škytavka,
- zvýšené množství jaterních enzymů v krvi.
Méně časté nežádoucí účinky (mohou postihnout až 1 ze 100 lidí) jsou:
- závrať, ospalost,
- akné, vyrážka,
- úzkost,
- říhání, pocit nevolnosti, zvracení, pálení žáhy, bolest žaludku, sucho v ústech, plynatost,
- častější, bolestivé nebo pálivé močení,
- slabost, celkový pocit nepohody,
- návaly horka/zarudnutí obličeje nebo kůže,
- rychlý nebo nepravidelný tep,
- horečka se zvýšeným rizikem infekce, snížení počtu červených krvinek.
Vzácné nežádoucí účinky (mohou postihnout až 1 z 1 000 lidí) jsou:
- potíže s přemýšlením, nedostatek energie, poruchy vnímání chutí,
- citlivost kůže na sluneční světlo, nadměrné pocení, mastná kůže, boláky na kůži, svědivá vyrážka, Stevens-Johnsonův syndrom/toxická epidermální nekrolýza (vzácná, závažná kožní reakce),
- euforie (pocit nadměrného štěstí), dezorientace,
- bakteriální infekce, plísňové infekce,
- těžká zácpa, žaludeční vředy, zánět tenkého střeva a tlustého střeva, boláky v ústech, nadýmání,
- časté močení, větší výdej moči, než je obvyklé, přítomnost cukru nebo krve v moči,
- pocit nepohody na hrudi, otok, změna způsobu chůze,
- kašel, hlen v zadní části hrdla (tzv. zadní rýma), podráždění hrdla, kýchání, bolest v krku,
- výtok z očí a svědění očí,
- zvonění v uších,
- svalové křeče, svalová slabost,
- nadměrná žízeň,
- pomalý srdeční tep, onemocnění srdce a cév,
- pokles počtu bílých krvinek, nízké hladiny sodíku v krvi, pokles tělesné hmotnosti.
Hlášení nežádoucích účinků
Pokud se u Vás nebo Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek EMEND uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Tobolku vyjímejte z blistru až těsně před užitím.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace
Co EMEND obsahuje
- Léčivou látkou je aprepitantum. Jedna 125mg tvrdá tobolka obsahuje aprepitantum 125 mg. Jedna 80mg tvrdá tobolka obsahuje aprepitantum 80 mg.
- Pomocnými látkami jsou: sacharóza, mikrokrystalická celulóza (E 460), hyprolóza (E 463), natrium-lauryl-sulfát, želatina, oxid titaničitý (E 171), šelak, hydroxid draselný a černý oxid železitý (E 172); 125mg tvrdá tobolka obsahuje také červený oxid železitý (E 172) a žlutý oxid železitý (E 172).
Jak EMEND vypadá a co obsahuje toto balení
125mg tvrdá tobolka je neprůhledná s růžovým víčkem a bílým tělem; na těle je po obvodu černým
inkoustem vytištěno „462“ a „125 mg“.
80mg tvrdá tobolka je neprůhledná s víčkem a tělem bílé barvy; na těle je po obvodu černým
inkoustem vytištěno „461“ a „80 mg“.
Tvrdé tobolky EMEND 125 mg a 80 mg se dodávají v následující velikosti balení:
- balení pro 3-denní léčbu obsahující jednu 125mg tobolku dvě 80mg tobolky
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
Výrobce
Merck Sharp & Dohme B. V Waarderweg 39 2031 BN Haarlem Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Eunrapnu
MepK fflapn h floyM Etnrapnn EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com
Česká republika
Merck Sharp & Dohme s.r.o.
Tel.: +420 233 010 111 dpoc_czechslovak@merck.com
Danmark
MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com
Lietuva
UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Magyarország
MSD Pharma Hungary Kft.
Tel.: +36 1 888 53 00 hungary_msd@merck.com
Malta
Merck Sharp & Dohme Cyprus Limited
Tel: 8007 4433 (+356 99917558) malta_info@merck.com
Deutschland
MSD SHARP & DOHME GMBH
Tel: 0800 673 673 673 (+49 (0) 89 4561 2612)
Eesti
Merck Sharp & Dohme OU Tel.:+372 6144 200 msdeesti@merck.com
EXXáSa
MSD A.OB.E.E.
TpA,: +30 210 98 97 300 dpoc_greece@merck.com
Espaňa
Merck Sharp & Dohme de Espana, S.A.
Tel: +34 91 321 06 00 msd_info@merck.com
France
MSD France
Tél: +33 (0) 1 46 40 40
Hrvatska
Merck Sharp & Dohme d.o.o.
Tel: + 385 1 6611 333 croatia_info@merck.com
Ireland
Merck Sharp & Dohme Ireland (Human Health) Limited
Tel: +353 (0)1 2998700 medinfo_ireland@merck.com
Ísland
Vistor hf.
Simi: +354 535 7000
Italia
MSD Italia S.r.l.
Tel: +39 06 361911 medicalinformation.it@merck.com
Kúnpoq
Merck Sharp & Dohme Cyprus Limited Tp^: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Nederland
Merck Sharp & Dohme BV
Tel: 0800 9999000 (+31 23 5153153)
Norge
MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no
Osterreich
Merck Sharp & Dohme Ges.m.b.H.
Tel: +43 (0) 1 26 044 msd-medizin@merck.com
Polska
MSD Polska Sp.z o.o.
Tel:+48 22 549 51 00 msdpolska@merck.com
Portugal
Merck Sharp & Dohme, Lda Tel:+351 21 446 5700 clic@merck.com
Románia
Merck Sharp & Dohme Romania S.R.L.
Tel:+ 4021 529 29 00 msdromania@merck.com
Slovenija
Merck Sharp & Dohme, inovativna zdravila d.o.o.
Tel:+ 386 1 5204 201 msd_slovenia@merck.com
Slovenská republika
Merck Sharp & Dohme, s. r. o.
Tel:+421 2 58282010 dpoc_czechslovak@merck.com
Suomi/Finland
MSD Finland Oy
Puh/Tel: +358 (0) 9 804 650
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Příbalová informace: informace pro uživatele
EMEND 125 mg prášek pro perorální suspenzi
aprepitantum
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje. Tato příbalová informace byla napsána pro rodiče nebo ošetřovatele, kteří budou tento lék dítěti podávat - přečtěte si ji prosím pečlivě.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se lékaře, lékárníka nebo zdravotní sestry.
- Tento přípravek byl předepsán výhradně Vašemu dítěti. Nedávejte jej žádné další osobě. Mohl
by jí ublížit, a to i tehdy, má-li stejné známky onemocnění.
- Pokud se u Vašeho dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je EMEND a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete EMEND podávat
3. Jak se EMEND užívá
4. Možné nežádoucí účinky
5 Jak EMEND uchovávat
6. Obsah balení a další informace
1. Co je EMEND a k čemu se používá
Přípravek EMEND obsahuje léčivou látku „aprepitant“. Ta patří do skupiny léčiv nazývaných "antagonisté receptoru neurokininu 1 (NK1)".
Mozek má zvláštní oblast, která řídí pocit nevolnosti a zvracení. Přípravek EMEND funguje tak, že blokuje signály do této oblasti, čímž omezuje pocit nevolnosti a zvracení.
Prášek pro perorální suspenzi se používá u dětí ve věku 6 měsíců až méně než 12 let v kombinaci s dalšími léčivy k předcházení nevolnosti a zvracení vyvolanými chemoterapií (léčbou rakoviny), která jsou silnými a středně silnými spouštěči pocitu nevolnosti a zvracení (jako je cisplatina, cyklofosfamid, doxorubicin nebo epirubicin).
2. Čemu musíte věnovat pozornost, než začnete EMEND podávat Neupodávejte EMEND
- jestliže je dítě alergické na aprepitant nebo na kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
- jestliže dítě užívá léky, které obsahují „pimozid” (na problémy s duševním zdravím).
- jestliže dítě užívá „terfenadin” nebo „astemizol“’ (na sennou rýmu a jiné alergie).
- jestliže dítě užívá „cisaprid“’ - (na zažívací problémy).
Tento lék nepodávejte, pokud se dítětě týká cokoli zvýše uvedeného a lékaře dítěte informujte, pokud dítě užívá kterýkoli z výše uvedených léků. To proto, že léčba bude muset být před nasazením tohoto léku upravena. Pokud si nejste jistý(á), poraďte se předtím, než podáte tento lék, s lékařem, lékárníkem nebo zdravotní sestrou.
Upozornění a opatření
Před podáním tohoto přípravku dítěti se poraďte s lékařem, lékárníkem nebo zdravotní sestrou.
Problémy s játry
Pokud Vaše dítě trpí onemocněním jater, řekněte to před zahájením léčby přípravkem EMEND lékaři. To proto, že játra hrají důležitou roli při rozkladu léku v těle. Proto lékař může během léčby potřebovat sledovat stav jater.
Děti a dospívající
Nepodávejte přípravek EMEND prášek pro perorální dětem mladším 6 měsíců nebo které váží méně než 6 kg, ani dospívajícím mezi 12 a 18 lety, protože prášek pro perorální suspenzi nebyl u této populace hodnocen.
Další léčivé přípravky a přípravek EMEND
Informujte lékaře, lékárníka nebo zdravotní sestru o všech lécích, které dítě užívá, které v nedávné době užívalo nebo které možná bude užívat. To proto, že přípravek EMEND může ovlivňovat účinky jiných léků během léčby přípravkem EMEND a po ní. Jiné léky mohou také ovlivňovat účinky tohoto léku.
Přípravek EMEND nepodávejte a informujte lékaře nebo lékárníka, pokud dítě užívá některý z následujících léků (viz také bod „Nepodávejte EMEND“). To proto, že jejich podávání se bude muset před zahájením podávání přípravku EMEND upravit:
- pimozid - na problémy s duševním zdravím
- terfenadin a astemizol - na sennou rýmu a jiné alergie
- cisaprid - na trávicí problémy
Tento lék nepodávejte a informujte lékaře nebo lékárníka, pokud se dítětě týká cokoli z výše uvedeného.
Informujte lékaře, lékárníka nebo zdravotní sestru, pokud dítě užívá kterýkoli z následujících léků:
- léky ovlivňující imunitní systém - jako je cyklosporin, takrolimus, sirolimus, everolimus
- alfentanil, fentanyl - proti bolesti
- chinidin - na nepravidelný tep srdce
- léky proti rakovině - jako je irinotekan, etoposid, vinorelbin, ifosfamid
- léky obsahující „deriváty námelových alkaloidů“ - jako je ergotamin a diergotamin - na migrénu
- léky na ředění krve - jako je warfarin, acenokumarol. Dítě může během léčby přípravkem EMEND potřebovat krevní testy.
- antibiotika k léčbě infekcí - jako je rifampicin, klarithromycin, telithromycin
- fenytoin - k léčbě záchvatů (křečí)
- karbamazepin - na deprese a epilepsii
- midazolam, triazolam, fenobarbital - utišující léky nebo léky usnadňující spánek
- třezalka tečkovaná - bylinný lék na deprese
- inhibitory proteázy - na infekce HIV
- ketokonazol, kromě šampónu (používá se k léčbě Cushingova syndromu - kdy tělo vytváří nadměrná množství kortizolu)
- protiplísňové léky, jako je itrakonazol, vorikonazol, posakonazol
- nefazodon - na deprese
- kortikosteroidy - jako je dexamethason a methylprednisolon
- léky proti úzkosti, jako je alprazolam
- tolbutamid - na cukrovku
- léky k zabránění početí, které mohou zahrnovat antikoncepční pilulky, kožní náplasti, implantáty a některá nitroděložní tělíska, která uvolňují hormony. Ty nemusí fungovat odpovídajícím způsobem, pokud se používají s tímto lékem. Během léčby tímto lékem a až 2 měsíce po ukončení léčby můžete potřebovat používat jiné nebo dodatečné nehormonální formy antikoncepce.
Pokud se dítěte něco z výše uvedeného týká (nebo si nejste jistý(á)), poraďte se předtím, než podáte tento lék, s lékařem, lékárníkem nebo zdravotní sestrou.
Těhotenství a kojení
Tento přípravek se během těhotenství a kojení nesmí užívat, pokud to není nezbytně nutné.
Ohledně informací týkajících se těhotenství, kojení a antikoncepce požádejte o radu svého lékaře. Řízení dopravních prostředků a obsluha strojů
Je nutno vzít v úvahu, že u některých lidí může po užití přípravku EMEND vzniknout pocit závrati a ospalosti. Pokud Vaše dítě má pocit závrati nebo ospalosti, nesmí po užití tohoto přípravku jezdit na kole ani používat žádné nástroje nebo obsluhovat stroje.
Přípravek EMEND obsahuje sacharózu a laktózu
Tobolky přípravku EMEND obsahují sacharózu a laktózu. Pokud Vás lékař řekl, že Vaše dítě nesnáší některé cukry, poraďte se s lékařem, než začnete tento přípravek dítěti podávat.
3. Jak se EMEND užívá
Zdravotničtí pracovníci: Viz pokyny pro zdravotnické pracovníky pro přípravu perorální suspenze na konci příbalové informace. Ty Vám řeknou jak připravit dávku přípravku EMEND prášek pro perorální suspenzi.
Rodiče a ošetřovatelé: Tento přípravek dítěti vždy podávejte přesně podle pokynů lékaře, lékárníka nebo zdravotní sestry. Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou.
Je velmi důležité podávat tento lék přesně podle pokynů níže.
Pro každou dávku přípravku EMEND dostanete předplněný perorální dávkovač obsahující předepsanou dávku pro dítě.
Uchovávejte dávkovač v chladničce (při teplotě 2 °C - 8 °C) předtím, než podáte lék dítěti.

Lék použijte během 2 dnů od obdržení od zdravotnického pracovníka.
Před podáním může být lék ponechán při pokojové teplotě (při teplotě do 30 °C) po dobu až 3 hodiny.

Barva léku v perorálním dávkovači může mít různé odstíny růžové (světle růžová až tmavě růžová). To je normální a lék je v pořádku k použítí.
- Sundejte uzávěr perorálního dávkovače.
- Umístěte špičku perorálního dávkovače do úst dítěte podél vnitřní strany tváře buď na pravé nebo levé straně.
- Pomalu tlačte na píst až dolů, aby byl podán všechen lék v perorálním dávkovači. Nemůže-li dítě užít celou dávku, volejte dětského lékaře.
Po ukončení podání nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
Kolik přípravku se podává
- Správnou dávku prášku pro perorální suspenzi vypočítá lékař na základě hmotnosti dítěte.
- Dávku neměňte ani léčbu neukončujte bez předchozí porady s lékařem, lékárníkem nebo zdravotní sestrou.
Kdy se přípravek užívá
1. den:
- tento lék podejte 1 hodinu před zahájením chemoterapeutického cyklu.
2. a 3. den:
- pokud se dítěti nepodává žádná chemoterapie - lék podejte ráno.
- pokud se dítěti podává chemoterapie - lék podejte 1 hodinu před zahájením chemoterapeutického cyklu.
Přípravek EMEND lze užívat bez ohledu na jídlo.
Tento přípravek vždy podávejte s dalšími léky, aby se zabránilo pocitu nevolnosti a zvracení. Po léčbě přípravkem EMEND může lékař požádat, aby dítě pokračovalo v užívání dalších léků k zabránění pocitu nevolnosti a zvracení, které mohou zahrnovat:
- kortikosteroid - jako je dexamethason a
- „antagonistu 5HT3“ - jako je ondansetron.
Pokud si nejste jistý(á), poraďte se s lékařem, lékárníkem nebo zdravotní sestrou.
Jestliže jste podal(a) více přípravku EMEND, než jste měl(a)
Nepodávejte dítěti více tohoto léku, než doporučil lékař. Pokud dítěti podáte více, než se mělo podat, ihned se obraťte na lékaře.
Jestliže jste zapomněl(a) přípravek EMEND podat
Pokud dítě vynechá dávku tohoto léku, poraďte se s lékařem.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky, může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
T-W r V r Vil r r v» 1
Závažné nežádoucí účinky
Přestaňte tento lék podávat a ihned navštivte lékaře, pokud Vy nebo dítě zaznamenáte kterýkoli z následujících závažných nežádoucích účinků - dítě může potřebovat neodkladnou lékařskou pomoc: - alergická reakce - příznaky mohou zahrnovat kopřivku, vyrážku, svědění, potíže s dýcháním nebo polykáním (četnost výskytu není známa).
Pokud zaznamenáte některý z výše uvedených závažných nežádoucích účinků, přestaňte tento lék podávat a ihned vyhledejte lékaře.
Dalš í nežádoucí účinky
Pokud Vy nebo dítě zaznamenáte kterýkoliv z následujících nežádoucích účinků, informujte lékaře, lékárníka nebo zdravotní sestru:
Časté: mohou postihnout až 1 z 10 lidí
- zácpa nebo poruchy trávení
- bolesti hlavy
- pocit únavy
- ztráta chuti k jídlu
- škytavka
- zvýšené množství jaterních enzymů v krvi (ukáže se v testech).
Méně časté: mohou postihnout až 1 ze 100 lidí
- pocit závratí nebo ospalosti
- akné, vyrážka
- pocit úzkosti
- říhání, pocit nevolnosti, zvracení, pálení žáhy, bolest žaludku, sucho v ústech, plynatost
- bolestivé nebo pálivé močení
- pocit slabosti, celkový pocit nepohody
- návaly horka/zarudnutí obličeje nebo kůže
- rychlý nebo nepravidelný tep
- horečka se zvýšeným rizikem infekce, snížení počtu červených krvinek (ukáže se v testech). Vzácné: mohou postihnout až 1 z 1 000 lidí
- potíže s přemýšlením, nedostatek energie, poruchy vnímání chutí
- citlivost kůže na sluneční světlo, nadměrné pocení, mastná kůže, boláky na kůži, svědivá vyrážka, Stevens-Johnsonův syndrom nebo toxická epidermální nekrolýza (vzácné, závažné kožní reakce)
- euforie (pocit nadměrného štěstí), pocit zmatenosti
- bakteriální infekce, plísňové infekce
- těžká zácpa, žaludeční vředy, zánět tenkého střeva a tlustého střeva, boláky v ústech, nadýmání
- časté močení nebo větší výdej moči, než je obvyklé, přítomnost cukru nebo krve v moči
- pocit nepohody na hrudi, otok, změna způsobu chůze
- kašel, hlen v zadní části hrdla (tzv. zadní rýma), podráždění hrdla, kýchání, bolest v krku
- výtok z očí a svědění očí
- zvonění v uších
- svalové křeče, svalová slabost
- nadměrná žízeň
- pomalý srdeční tep, onemocnění srdce a cév
- nízký počet bílých krvinek, nízké hladiny sodíku v krvi, pokles tělesné hmotnosti.
Hlášení nežádoucích účinků
Pokud se u dítěte vyskytne kterýkoli z nežádoucích účinků, sdělte to lékaři, lékárníkovi nebo zdravotní sestře. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek EMEND uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí. Před rekonstitucí:
Přípravek Emend bude obecně uchováván zdravotnickými praovníky. Podrobnosti o uchovávání, pokud je budete potřebovat, jsou následující:
Nepodávejte dítěti tento přípravek po uplynutí doby použitelnosti, uvedené na krabičce a sáčku za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Tento léčivý přípravek nevyžaduje žádné zvláštní teplotní podmínky uchovávání.
Uchovávejte v původním obalu, aby byl přípravek chráněn před vlhkostí.
Po rekonstituci:
Před podáním může být perorální suspenze uchovávána při pokojové teplotě po dobu až 3 hodiny (při teplotě do 30 °C). Také může být uchovávána v chladničce po dobu až 72 hodin (při teplotě 2 °C -8 °C).
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co EMEND obsahuje
- Léčivou látkou je aprepitantum. Jeden sáček obsahuje aprepitantum 125 mg. Po rekonstituci obsahuje 1 ml perorální suspenze aprepitantum 25 mg.
- Pomocnými látkami jsou: hyprolóza (E 463), natrium-lauryl-sulfát (E 487), sacharóza a laktóza (viz bod 2, odstavec „Přípravek EMEND obsahuje sacharózu a laktózu“), červený oxid železitý (E 172) a natrium-stearyl-fumarát (E 485).
Jak EMEND vypadá a co obsahuje toto balení
Prášek pro perorální suspenzi je růžový až světle růžový prášek v sáčku na jedno použití.
Krabička na jedno použití.
Jedna krabička obsahuje jeden sáček, jeden 1ml a jeden 5ml perorální dávkovač (polypropylenový se silikonovým o-kroužkem), jeden uzávěr a jednu mísicí nádobku (polypropylenovou).
Držitel rozhodnutí o registraci a výrobce
Držitel rozhodnutí o registraci Merck Sharp & Dohme Ltd. Hertford Road, Hoddesdon Hertfordshire EN 11 9BU Velká Británie
Výrobce
Merck Sharp & Dohme B. V Waarderweg 39 2031 BN Haarlem Nizozemsko
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci:
Lietuva
UAB Merck Sharp & Dohme Tel.:+370 5 278 02 47 msd_lietuva@merck.com
Luxembourg/Luxemburg
MSD Belgium BVBA/SPRL Tél/Tel: +32(0)27766211 dpoc_belux@merck.com
Belgique/Belgie/Belgien
MSD Belgium BVBA/SPRL
Tél/Tel: 0800 38 693 (+32(0)27766211)
Eunrapnu
MepK fflapn h floyM Etnrapnn EOOfl Ten.:+359 2 819 3737 info-msdbg@merck.com
|
Česká republika Merck Sharp & Dohme s.r.o. Tel.: +420 233 010 111 dpoc_czechslovak@merck.com |
Magyarország MSD Pharma Hungary Kft. Tel.: +36 1 888 53 00 hungary_msd@merck.com |
|
Danmark MSD Danmark ApS Tlf: + 45 4482 4000 dkmail@merck.com |
Malta Merck Sharp & Dohme Cyprus Limited Tel: 8007 4433 (+356 99917558) malta_info@merck.com |
|
Deutschland MSD SHARP & DOHME GMBH Tel: 0800 673 673 673 (+49 (0) 89 4561 2612) |
Nederland Merck Sharp & Dohme BV Tel: 0800 9999000 (+31 23 5153153) |
|
Eesti Merck Sharp & Dohme OU Tel.:+372 6144 200 msdeesti@merck.com |
Norge MSD (Norge) AS Tlf: +47 32 20 73 00 msdnorge@msd.no |
|
EXXáSa MSD A.OB.E.E. Tr^: +30 210 98 97 300 dpoc_greece@merck.com |
Osterreich Merck Sharp & Dohme Ges.m.b.H. Tel: +43 (0) 1 26 044 msd-medizin@merck.com |
|
Espaňa Merck Sharp & Dohme de Espana, S.A. Tel: +34 91 321 06 00 msd_info@merck.com |
Polska MSD Polska Sp. z o.o. Tel:+48 22 549 51 00 msdpolska@merck.com |
|
France MSD France Tél: +33 (0) 1 46 40 40 |
Portugal Merck Sharp & Dohme, Lda Tel:+351 21 446 5700 clic@merck.com |
|
Hrvatska Merck Sharp & Dohme d.o.o. Tel: + 385 1 6611 333 croatia info@merck.com |
Románia Merck Sharp & Dohme Romania S.R.L. Tel:+ 4021 529 29 00 msdromania@merck.com |
|
Ireland Merck Sharp & Dohme Ireland (Human Health) Limited Tel: +353 (0)1 2998700 medinfo ireland@merck.com |
Slovenija Merck Sharp & Dohme, inovativna zdravila d.o.o. Tel:+ 386 1 5204 201 msd_slovenia@merck.com |
|
Ísland Vistor hf. Simi: +354 535 7000 |
Slovenská republika Merck Sharp & Dohme, s. r. o. Tel:+421 2 58282010 dpoc_czechslovak@merck.com |
|
Italia MSD Italia S.r.l. Tel: +39 06 361911 medicalinformation.it@merck.com |
Suomi/Finland MSD Finland Oy Puh/Tel: +358 (0) 9 804 650 |
Kúrcpog
Merck Sharp & Dohme Cyprus Limited T^: 800 00 673 (+357 22866700) cyprus_info@merck.com
Latvija
SIA Merck Sharp & Dohme Latvija Tel: +371 67364 224 msd_lv@merck.com
Sverige
Merck Sharp & Dohme (Sweden) AB Tel: +46 77 5700488 medicinskinfo@merck.com
United Kingdom
Merck Sharp & Dohme Limited Tel: +44 (0) 1992 467272 medicalinformationuk@merck.com
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Instrukce pro zdravotnické pracovníky k přípravě perorální suspenze
Jedno balení přípravku EMEND obsahuje sáček s práškem pro perorální suspenzi, 1ml a 5ml perorální dávkovač, jeden uzávěr a jednu mísicí nádobku.

uzávěr
1.
Naplňte mísicí nádobku pitnou vodou o pokojové teplotě.

2.
Naplňte 5ml perorální dávkovač 4,6 ml vody z mísicí nádobky.
Ujistěte se, že perorální dávkovač neobsahuje vzduch (je-li vzduch přítomen, odstraňte jej).

3.
Zlikvidujte všechnu zbývající vodu v mísicí nádobce.

4.
Dejte 4,6 ml vody z perorálního dávkovače zpět do mísicí nádobky.

5.
Jeden sáček přípravku EMEND pro přípravu perorální suspenze obsahuje 125 mg aprepitantu, který se rozpustí ve 4,6 ml vody, čímž se získá finální koncentrace 25 mg/ml.
Držte sáček přípravku EMEND prášek pro perorální suspenzi svisle a setřepejte obsah do spodní části, než jej otevřete.
6. Vysypte veškerý obsah sáčku do 4,6 ml vody v mísicí nádobce a uzavřete víčkem.
Odtrhněte

7. Suspenzi přípravku EMEND j emným kroužením 20krát promíchejte; potom opatrně mísicí nádobku 5krát obraťte.
Abyste zabránil(a) pěnění, mísicí nádobkou netřepejte. Směs bude zakalená, růžová až světle růžová.

8. Zkontrolujte přítomnost shluků a pěny ve směsi přípravku EMEND:
- Jsou-li přítomny nějaké shluky, opakujte krok 7, dokud shluky nezmizí.
- Je-li přítomna pěna, počkejte, než pěna zmizí, než přejdete ke kroku 9.

|
Tělesná hmotnost |
Objem dávky perorálně podávané suspenze | ||
|
1. den |
2. den |
3. den | |
|
méně než 6 kg |
Nedoporučuje se | ||
|
6 kg až méně než 8 kg |
1 ml (25 mg) |
0,6 ml (15 mg) |
0,6 ml (15 mg) |
|
8 kg až méně než 10 kg |
1,2 ml (30 mg) |
0,8 ml (20 mg) |
0,8 ml (20 mg) |
|
10 kg až méně než 12 kg |
1,4 ml (35 mg) |
1 ml (25 mg) |
1 ml (25 mg) |
|
12 kg až méně než 15 kg |
1,8 ml (45 mg) |
1,2 ml (30 mg) |
1,2 ml (30 mg) |
|
15 kg až méně než 20 kg |
2,4 ml (60 mg) |
1,6 ml (40 mg) |
1,6 ml (40 mg) |
|
20 kg až méně než 25 kg |
3 ml (75 mg) |
2 ml (50 mg) |
2 ml (50 mg) |
|
25 kg až méně než 30 kg |
3,6 ml (90 mg) |
2,4 ml (60 mg) |
2,4 ml (60 mg) |
|
30 kg a více |
Obsah (125 mg) mísicí nádobky natáhněte do perorálního dávkovače (přibližně 5 ml) |
3,2 ml (80 mg) |
3,2 ml (80 mg) |
|
9. Naplňte perorální dávkovač předepsanou dávkou z mísicí nádobky v souladu s tabulkou výše. - Na základě dávky zvolte perorální dávkovač: - Jestliže je dávka 1 ml a nižší, použijte 1ml perorální dávkovač. - Jestliže je dávka vyšší než 1 ml, použijte 5ml perorální dávkovač. - Je obvyklé mít v mísicí nádobce přebytek léčiva. Ujistěte se, že perorální dávkovač neobsahuje vzduch (je-li vzduch přítomen, odstraňte jej). Ujistěte se, že perorální dávkovač obsahuje předepsanou dávku. |
1ml perorální dávkovač |
NEBO 5ml perorální dávkovač |
|
10. Umístěte uzávěr na perorální dávkovač, dokud nezapadne. 11. Pokud není dávka podána bezprostředně po odměření, uchovávejte naplněný(é) perorální dávkovač(e) v chladničce při teplotě 2 °C - 8 °C po dobu až 72 hodin před použitím. Při výdeji dávky (dávek) ošetřovateli (ošetřovatelce) jej (ji) informujte, aby perorální dávkovač(e) uchovával(a) v chladu do doby, než bude připraven(a) dávku podat. 12. Perorální suspenze může být uchovávána při pokojové teplotě (při teplotě do 30 °C) po dobu až 3 hodiny před použitím. | ||
|
Zlikvidujte všechnu zbývající směs a odpadní materiál. Veškerý nepoužitý léčivý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky. | ||
131