Volibris 10 Mg
SOUHRN ÚDAJŮ O PŘÍPRAVKU
PŘÍLOHA I
Volibris 5 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje ambrisentanum 5 mg.
Pomocné látky se známým účinkem:
Jedna tableta obsahuje přibližně 95 mg laktosy (ve formě monohydrátu), přibližně 0,25 mg sójového lecithinu (E322) a přibližně 0,11 mg hlinitého laku červeně Allura AC (E129).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Světle růžové, čtvercovité, vypouklé potahované tablety s vyraženým "GS" na jedné straně a "K2C" na straně druhé.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Volibris je indikován k léčbě plicní arteriální hypertenze (PAH) II. až III. funkční třídy (FC) dle klasifikace WHO u dospělých pacientů včetně použití v kombinované terapii (viz bod 5.1). Účinnost byla prokázána u idiopatické PAH (IPAH) a PAH spojené s onemocněním pojivové tkáně.
4.2 Dávkování a způsob podání
Léčba musí být zahájena lékařem, který má zkušenosti s léčbou PAH.
Dávkování
Ambrisentan v monoterapii
Volibris se užívá perorálně v počáteční dávce 5 mg jedenkrát denně a dávka může být zvýšena na 10 mg denně v závislosti na klinické odpovědi a snášenlivosti.
Ambrisentan v kombinaci s tadalafilem
Při užívání v kombinaci s tadalafilem má být Volibris titrován do dávky 10 mg jednou denně.
Ve studii AMBITION pacienti užívali 5 mg ambrisentanu denně po dobu prvních 8 týdnů před zvýšením až na 10 mg, v závislosti na snášenlivosti (viz bod 5.1). Při užívání v kombinaci s tadalafilem, pacienti začínali s 5 mg ambrisentanu a 20 mg tadalafilu. V závislosti na snášenlivosti byla dávka tadalafilu zvýšena po 4 týdnech na 40 mg a dávka ambrisentanu byla zvýšena po 8 týdnech na 10 mg. Tohoto dosáhlo více než 90 % pacientů. Dávky také mohly být sníženy v závislosti na snášenlivosti.
Na základě omezených údajů lze usuzovat, že náhlé přerušení léčby ambrisentanem není spojeno s opětovným zhoršením PAH.
Pokud je ambrisentan podáván společně s cyklosporinem A, měla by být jeho dávka omezena na 5 mg jednou denně a pacient by měl být pečlivě monitorován (viz bod 4.5 a 5.2).
Zvláštní populace
Starší pacienti
U pacientů starších 65 let není nutná žádná úprava dávkování (viz bod 5.2).
Pacienti s _poruchou renální _funkce
U pacientů s poruchou renální funkce není nutná žádná úprava dávkování (viz bod 5.2). U jedinců s těžkou poruchou renální funkce (clearance kreatininu <30 ml/min) jsou s podáváním ambrisentanu jen omezené zkušenosti; proto u této skupiny pacientů musí být léčba zahajována s opatrností a zvláštní opatrnost je nezbytná zejména při zvýšení dávky ambrisentanu na 10 mg.
Pacienti s _poruchou hepatální _funkce
Ambrisentan nebyl hodnocen u jedinců s poruchou funkce jater (s cirhózou nebo bez cirhózy). Vzhledem k tomu, že hlavní metabolickou cestou ambrisentanu je glukuronidace a oxidace s následným vylučováním žlučí, lze předpokládat, že jaterní poškození může zvýšit expozici (Cmax a AUC) ambrisentanu. Léčba ambrisentanem se nesmí zahajovat u pacientů s těžkým jaterním poškozením nebo s klinicky významně zvýšenými hodnotami jaterních aminotransferáz (více než trojnásobek horního limitu normálních hodnot (>3x ULN); viz body 4.3 and 4.4).
Pediatrická populace
Bezpečnost a účinnost ambrisentanu u dětí a dospívajících mladších 18 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Doporučuje se užívat tablety celé, nalačno nebo s jídlem. Doporučuje se, aby tableta nebyla dělena, drcena ani žvýkána.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku, sóju nebo kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
• Těhotenství (viz bod 4.6).
• Ženy v reprodukčním věku, které nepoužívají účinnou antikoncepci (viz body 4.4 a 4.6).
• Kojení (viz bod 4.6).
• Těžké jaterní poškození (s cirhózou nebo bez cirhózy) (viz bod 4.2).
• Výchozí hodnoty jaterních aminotransferáz (aspartátaminotransferázy (AST) a/nebo alaninaminotransferázy (ALT) >3x ULN (viz body 4.2 and 4.4).
• Idiopatická plicní fibróza (IPF) se sekundární plicní hypertenzí nebo bez ní (viz bod 5.1).
4.4 Zvláštní upozornění a opatření pro použití
Ambrisentan nebyl hodnocen u dostatečného množství pacientů, aby bylo možno určit poměr prospěchu/rizika u pacientů s PAH I. funkční třídy dle klasifikace WHO.
U pacientů s PAH IV. funkční třídy dle klasifikace WHO nebyla účinnost ambrisentanu v monoterapii hodnocena. Při zhoršení klinického stavu by měla být zvážena léčba, která se doporučuje při těžkém stadiu tohoto onemocnění (např. epoprostenol).
Jaterní funkce
S PAH bývají spojeny poruchy jaterní ch funkcí. Při léčbě ambrisentanem byly zaznamenány případy odpovídající autoimunitní hepatitidě, včetně možné exacerbace již existující autoimunitní hepatitidy, poškození jater a zvýšené hodnoty jaterních enzymů vznikající v možné souvislosti s léčbou (viz body 4.8 a 5.1). Před zahájením léčby ambrisentanem by proto měly být posouzeny jaterní aminotransferázy (ALT a AST) a léčba nesmí být zahájena u pacientů s výchozími hodnotami ALT a/nebo AST >3x ULN (viz bod 4.3).
Pacienti by měli být sledováni s ohledem na příznaky poškození jater a doporučuje se monitorovat hodnoty ALT a AST každý měsíc. Pokud dojde u pacienta k rozvoji přetrvávajícího, nejasného, klinicky významného zvýšení hodnot ALT a/nebo AST nebo je-li zvýšení hodnot ALT a/nebo AST spojeno se známkami nebo příznaky jaterního poškození (např. žloutenka), má být terapie ambrisentanem přerušena.
U pacientů bez klinických příznaků jaterního poškození nebo žloutenky může být po úpravě hodnot jaterních enzymů zváženo opětovné zahájení léčby ambrisentanem. Doporučuje se konzultace s hepatologem.
Koncentrace hemoglobinu
Při léčbě antagonisty receptorů pro endotelin (ERA) včetně ambrisentanu byly zaznamenány snížené koncentrace hemoglobinu a snížení hematokritu. K tomuto snížení došlo většinou v průběhu prvních 4 týdnů léčby a později se hodnoty hemoglobinu obvykle stabilizovaly. Střední snížení koncentrace hemoglobinu od výchozích hodnot (v rozmezí od 0,9 do 1,2 g/dl) přetrvávalo po dobu až 4 let během léčby ambrisentanem v dlouhodobém otevřeném rozšíření pivotních klinických studiích fáze 3. V období po uvedení přípravku na trh byly hlášeny případy anémie vyžadující krevní transfuzi (viz bod 4.8).
U pacientů s klinicky významnou anémií se zahájení léčby ambrisentanem nedoporučuje. Doporučuje se, aby byly v průběhu léčby ambrisentanem hodnoty hemoglobinu a/nebo hematokritu monitorovány, např. v 1. měsíci, ve 3. měsíci a dále v pravidelných intervalech v souladu s klinickou praxí. Pokud je zaznamenán klinicky významný pokles hemoglobinu nebo hematokritu a jiné příčiny tohoto poklesu jsou vyloučeny, mělo by být zváženo snížení dávky nebo přerušení léčby. Výskyt anémie se zvýšil, když byl ambrisentan podáván v kombinaci s tadalafilem (frekvence výskytu nežádoucích účinků 15 %) v porovnání s výskytem anémie, kdy byly ambrisentan a tadalafil podávány samostatně jako monoterapie (7 % a 11 %).
Retence tekutin
Při užívání antagonistů receptorů pro endotelin (ERA) včetně ambrisentanu byl zaznamenán periferní edém. Periferní edém zaznamenaný v klinických studiích s ambrisentanem byl ve většině případů mírný až středně závažný, je však možné, že se vyskytoval častěji a že byl závažnější u pacientů >65 let. Periferní edém byl hlášen častěji při užívání dávky 10 mg ambrisentanu v krátkodobých klinických studiích (viz bod 4.8).
Po uvedení přípravku na trh byly hlášeny případy retence tekutin, ke kterým došlo během několika týdnů po zahájení léčby ambrisentanem. V některých případech bylo k obnovení rovnováhy tekutin nebo u dekompenzovaného srdečního selhání nutné podání diuretik nebo hospitalizace. Pokud je u pacientů zaznamenána již existující zvýšená retence tekutin, je třeba ji vhodným způsobem řešit ještě před zahájením léčby ambrisentanem.
Dojde-li během léčby ambrisentanem k rozvoji klinicky významné retence tekutin (s nárůstem tělesné hmotnosti nebo bez něj), je nutno posoudit příčinu tohoto stavu, kterou může být léčba ambrisentanem nebo již existující srdeční selhávání, a je třeba zvážit možnost eventuální specifické léčby nebo přerušení léčby ambrisentanem. Výskyt periferního edému se zvýšil, když byl ambrisentan podáván v kombinaci s tadalafilem (frekvence výskytu nežádoucího účinků 45 %), v porovnání s výskytem periferního edému, kdy byly ambrisentan a tadalafil podávány jako monoterapie (38 % a 28 %). Výskyt periferního edému byl nejvyšší během prvního měsíce po zahájení léčby.
Ženy v reprodukčním věku
Léčba přípravkem Volibris může být u žen v reprodukčním věku zahájena pouze v případě, že je výsledek jejich těhotenského testu provedeného před zahájením léčby negativní a pouze při používání účinné antikoncepce. V případě pochybností, jakou antikoncepci doporučit konkrétní pacientce, by měla být zvážena konzultace s gynekologem. V průběhu léčby ambrisentanem se doporučuje provádět jednou měsíčně těhotenské testy (viz body 4.3 a 4.6).
Plicní venookluzivní nemoc
U pacientů s plicní venookluzivní nemocí byly při užívání vazodilatačních léčivých přípravků, jako jsou například ERA, hlášeny případy vzniku edému plic. Tudíž dojde-li při léčbě ambrisentanem u pacientů s PAH ke vzniku akutního edému plic, musí být zvažována možnost přítomnosti venookluzivní nemoci.
Současné podávání s jinými léčivými přípravky
Pacienti, kterým je podáván ambrisentan, musí být po zahájení léčby rifampicinem pečlivě sledováni (viz bod 4.5 a 5.2).
Pomocné látky
Tablety přípravku Volibris obsahují laktosu. Tento přípravek by neměli užívat pacienti se vzácnými dědičnými problémy s intolerancí galaktosy, vrozeným deficitem laktasy nebo malabsorpcí glukosy a galaktosy.
Tablety přípravku Volibris obsahují azobarvivo hlinitý lak červeně Allura AC (E129), které může vyvolat alergickou reakci.
Tablety přípravku Volibris obsahují lecithin získaný ze sóji. Pokud je pacient přecitlivělý na sóju, nesmí ambrisentan užívat (viz bod 4.3).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Ambrisentan neinhibuje ani neindukuje enzymy I. fáze nebo II. fáze metabolické přeměny léčiva v klinicky významných koncentracích v předklinických studiích in vitro a in vivo. Z této skutečnosti lze usuzovat na nízký potenciál ambrisentanu k ovlivnění profilu léčivých přípravků metabolizovaných touto cestou.
Možná indukce aktivity CYP3A4 způsobená ambrisentanem byla hodnocena u zdravých dobrovolníků. Výsledky tohoto hodnocení nenasvědčují tomu, že by měl ambrisentan indukční účinek na izoenzym CYP3A4.
Cvklosporin A
Společné podávání ambrisentanu a cyklosporinu A u zdravých dobrovolníků vedlo v rovnovážném stavu ke dvojnásobnému zvýšení expozice ambrisentanu . To může být v důsledku cyklosporinem A způsobené inhibice transportérů a metabolických enzymů zahrnutých do farmakokinetiky ambrisentanu. Proto by dávka ambrisentanu měla být omezena na 5 mg jednou denně, pokud je podáván společně s cyklosporinem A (viz bod 4.2). Mnohočetné dávky ambrisentanu neměly na expozici cyklosporinu A vliv, a proto není úprava dávky cyklosporinu A nutná.
Rifampicin
Současné podávání rifampicinu (inhibitor transportního proteinu pro organické anionty [OATP], silný induktor CYP3A a 2C19 a induktor P-gp a uridindifosfát glukuronosyltransferáz [UGT]) s ambrisentanem bylo spojeno s přechodným (přibližně dvojnásobným) zvýšením expozice ambrisentanu po počátečních dávkách podaných zdravým dobrovolníkům. Do osmého dne se však hladina upravila a podávání rifampicinu v ustáleném stavu nemělo žádný klinicky relevantní vliv na expozici ambrisentanu. Pacienti, kterým je podáván ambrisentan, musí být po zahájení léčby rifampicinem pečlivě sledováni (viz body 4.5 a 5.2).
Inhibitory fosfodiesterázy
Současné podávání ambrisentanu s inhibitory fosfodiesterázy, a to se sildenafilem nebo tadalafilem (obě látky jsou substráty pro CYP3A4) zdravým dobrovolníkům nemělo významný vliv na farmakokinetiku inhibitorů fosfodiesterázy nebo ambrisentanu (viz bod 5.2).
Další cílená léčba PAH
Účinnost a bezpečnost ambrisentanu při současné další léčbě PAH (např. prostanoidy a stimulátory rozpustné guanylátcyklázy) nebyly v kontrolovaných klinických studiích u pacientů s PAH specificky hodnoceny (viz bod 5.1). Žádné specifické lékové interakce se stimulátory rozpustné guanylátcyklázy nebo prostanoidy se neočekávají na základě známých dat o biotransformaci (viz bod 5.2). Nicméně, studie specifických lékových interakcí s těmito léky nebyly provedeny.
Proto se při současném užívání těchto léčiv doporučuje postupovat s opatrností.
Perorální antikoncepce
V klinické studii se zdravými dobrovolníky neovlivnilo podávání ambrisentanu v dávce 10 mg jedenkrát denně v ustáleném stavu významně farmakokinetiku složek kombinované perorální antikoncepce - ethinylestradiolu a norethisteronu - po jednorázovém podání (viz bod 5.2). Na základě této farmakokinetické studie se nepředpokládá, že by ambrisentan významně ovlivňoval expozici antikoncepčním přípravkům s obsahem estrogenu nebo progestinu.
Warfarin
Ve studii se zdravými dobrovolníky neměl ambrisentan žádný vliv na farmakokinetiku warfarinu v ustáleném stavu ani na antikoagulační aktivitu warfarinu (viz bod 5.2). Podobně warfarin neměl klinicky signifikantní vliv na farmakokinetiku ambrisentanu. Navíc u pacientů neměl ambrisentan celkově žádný vliv na týdenní dávku antikoagulancií typu warfarinu, protrombinový čas (PT) ani na mezinárodní normalizovaný poměr (INR).
Ketokonazol
Podávání ketokonazolu (silného inhibitoru CYP3A4) v rovnovážném stavu nevedlo ke klinicky významnému zvýšení expozice ambrisentanu (viz bod 5.2).
Vliv ambrisentanu na xenobiotické transportéry
In vitro ambrisentan při klinicky relevantních koncentracích u lidí neinhibuje transportéry, včetně P-glykoproteinu (Pgp), proteinu resistence karcinomu prsu (BCRP), izoformy 2 proteinu mnohočetné lékové rezistence (MRP2), exportní pumpu žlučových kyselin (BSEP), polypeptidy transportující organické ionty (OATP1B1 and OATP1B3) a na sodíku závislý kotransportér taurocholátu sodného (NTCP).
Ambrisentan je substrátem pro eflux zprostředkovaný Pgp.
In vitro studie prováděné na potkaních hepatocytech taktéž prokázaly, že ambrisentan neindukuje expresi proteinů Pgp, BSEP ani MRP2 .
Podávání ambrisentanu nemělo v ustáleném stavu žádný klinicky významný účinek na farmakokinetiku digoxinu (substrátu Pgp) po jeho jednorázovém podání (viz bod 5.2).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Léčba ambrisentanem může být u žen ve fertilním věku zahájena pouze v případě, že je výsledek jejich těhotenského testu provedeného před zahájením léčby negativní a pouze při používání účinné antikoncepce. V průběhu léčby přípravkem Volibris se doporučuje provádět jednou měsíčně těhotenské testy.
Ambrisentan je v těhotenství kontraindikován (viz bod 4.3). Ve studiích na zvířatech byla prokázána teratogenita ambrisentanu. S podáváním u lidí nejsou žádné zkušenosti.
Ženy užívající ambrisentan musí být upozorněny na riziko poškození plodu a pokud dojde k otěhotnění, musí být zahájena alternativní léčba (viz body 4.3, 4.4 a 5.3).
Kojení
Není známo, zda je ambrisentan vylučován do lidského mateřského mléka. Vylučování ambrisentanu do mléka nebylo u zvířat hodnoceno. Z tohoto důvodu je u pacientek užívajících ambrisentan kojení kontraindikováno (viz bod 4.3).
Fertilita u mužů
Dlouhodobé podávání antagonistů receptorů pro endotelin (včetně ambrisentanu) u samců laboratorních zvířat bylo spojeno s rozvojem tubulární atrofie varlat (viz bod 5.3). Ačkoli nebyl ve studii ARIES-E nalezen žádný zřejmý důkaz škodlivého vlivu dlouhodobé expozice ambrisentanu na počet spermií, chronické podávání ambrisentanu bylo spojováno se změnami markerů spermatogeneze. Bylo pozorováno snížení plazmatických koncentrací inhibinu B a zvýšení plazmatických koncentrací FSH. Vliv na fertilitu u mužů není znám, ale narušení spermatogeneze nelze vyloučit. V klinických studiích nebylo dlouhodobé užívání ambrisentanu spojeno se změnami plazmatických hladin testosteronu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Ambrisentan má mírný nebo středně významný vliv na schopnost řídit a obsluhovat stroje. Při zvažování schopnosti pacienta provádět činnosti, které vyžadují rozhodovací, motorické a kognitivní schopnosti je třeba vzít v úvahu klinický stav pacienta a profil nežádoucích účinků ambrisentanu (jako je hypotenze, závrať, asténie a únava) (viz bod 4.8). Pacienti by měli být před řízením a obsluhou strojů upozorněni, jak je může ambrisentan ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost ambrisentanu jako monoterapie a/nebo v kombinaci byla hodnocena v klinických studiích s více než 1200 pacienty s PAH (viz bod 5.1). Nežádoucí účinky vycházející z údajů 12týdenní placebem kontrolované klinické studie, seřazené podle tříd orgánových systémů a četnosti, jsou zahrnuty níže. Informace z dlouhodobých, placebem nekontrolovaných studií (ARIES-E a AMBITION (kombinace s tadalafilem)), jsou též uvedeny níže. Žádné dříve nepozorované nežádoucí účinky nebyly při dlouhodobé léčbě nebo při kombinaci ambrisentanu s tadalafilem identifikovány. Bezpečnostní profil byl při delším pozorování vycházejícím z nekontrolovaných studií (průměrná doba sledování 79 týdnů) podobný bezpečnostnímu profilu vycházejícímu z krátkodobých studií. Údaje získané z rutinního farmakovigilančního sledování jsou rovněž uvedeny.
Nejčastěji pozorovanými nežádoucími účinky v souvislosti s léčbou ambrisentanem byly periferní edém, retence tekutin a bolest hlavy (zahrnující bolest hlavy způsobenou onemocněním vedlejších nosních dutin a migrénu). Vyšší dávky (10 mg) byly spojovány s vyšší incidencí těchto nežádoucích účinků a periferní edém měl u pacientů starších 65 let závažnější projevy v krátkodobých klinických studiích (viz bod 4.4).
Seznam nežádoucích účinků v tabulce
Skupiny četností jsou definovány následujícím způsobem: velmi časté (>1/10), časté (> 1/100 až <1/10), méně časté (> 1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit). U nežádoucích účinků závislých na dávce je uváděna skupina četností odpovídající vyšší dávce ambrisentanu. Skupiny četností neodrážejí další faktory, jako jsou rozdílná délka studií, zdravotní stav a laboratorní parametry pacienta před léčbou. Skupiny četností nežádoucích účinků určené na základě zkušeností z klinických studií nemusejí odrážet četnost nežádoucích účinků, které se objeví v průběhu běžné klinické praxe. V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Ambrisentan |
Ambrisentan |
Kombinace s | |
|
(ARIES-C a po |
(AMBITION a |
tadalafilem | |
|
uvedení na trh) |
ARIES-E) |
(AMBITION) | |
|
Poruchy krve a lymfatického systému | |||
|
Anémie (pokles hemoglobinu, pokles hematokritu) |
Časté1 |
Velmi časté |
Velmi časté |
|
Poruchy imunitního systému | |||
|
Reakce přecitlivělosti (např. angioedém, vyrážka, pruritus) |
Méně časté |
Časté |
Časté |
|
Poruchy nervového systému | |||
|
Bolest hlavy (včetně bolesti dutin v hlavě, migréna) |
Velmi časté2 |
Velmi časté |
Velmi časté |
|
Závrať |
Časté3 |
Velmi časté |
Velmi časté |
|
Oční poruchy | |||
|
Rozmazané vidění, poruchy zraku |
Není známo4 |
Časté |
Časté |
|
Poruchy ucha a ušního labyrintu | |||
|
Tinnitus |
NH |
NH |
Časté |
|
Náhlá ztráta sluchu |
NH |
NH |
Méně časté |
|
Srdeční poruchy | |||
|
Srdeční selhání |
Časté5 |
Časté |
Časté |
|
Palpitace |
Časté |
Velmi časté |
Velmi časté |
|
Cévní poruchy | |||
|
Časté3 |
Časté |
Časté | |
|
Návaly horka/zrudnutí |
Časté |
Časté |
Velmi časté |
|
Synkopa |
Méně časté3 |
Časté |
Časté |
|
Respirační, hrudní a mediastinální poruchy | |||
|
Epistaxe |
Časté3 |
Časté |
Časté |
|
Časté3,6 |
Velmi časté |
Velmi časté | |
|
Kongesce horních cest dýchacích (včetně nosu a vedlejších nosních dutin), sinusitida, nazofaryngitida, rinitida |
Časté7 | ||
|
Nazofaryngitida |
Velmi časté |
Velmi časté | |
|
Sinusistida, rinitida |
Časté |
Časté | |
|
Ucpaný nos |
Velmi časté |
Velmi časté | |
|
Gastrointestinální poruchy | |||
|
Časté3 | |||
|
Velmi časté |
Velmi časté | ||
|
Časté |
Velmi časté | ||
|
Velmi časté |
Velmi časté | ||
|
Časté |
Časté |
Časté | |
|
Zácpa |
Časté |
Časté |
Časté |
|
Poruchy jater a žlučových cest | |||
|
Poškození jater (viz bod 4.4) |
Méně časté3,8 |
NH |
NH |
|
Autoimunitní hepatitida (viz bod 4.4) |
Méně časté3,8 |
NH |
NH |
|
Zvýšení jaterních transamináz |
Časté3 |
NH |
NH |
|
Poruchy kůže a podkožní tkáně | |||
|
NH |
Časté9 |
Velmi časté9 | |
|
Celkové a jinde nezařazené poruchy a lokální reakce po podání | |||
|
Periferní otok, retence tekutin |
Velmi časté |
Velmi časté |
Velmi časté |
|
Bolest/nepříjemný pocit na hrudi |
Časté |
Časté |
Velmi časté |
|
Astenie |
Časté3 |
Časté |
Časté |
|
Únava |
Časté3 |
Velmi časté |
Velmi časté |
NH - nebylo hlášeno
1 Viz bod „Popis vybraných nežádoucích účinků".
2 Bolesti hlavy se objevují častěji při užívání ambrisentanu v dávce 10 mg.
3 Údaje získané z rutinního farmakovigilančního sledování a četnosti založené na zkušenostech z placebem kontrolovaných klinických studií.
4 Data odvozená z rutinního farmakovigilančního sledování
5 Většina hlášených případů srdečního selhání byla spojována s retencí tekutin. Údaje odvozené z rutinního farmakovigilančního sledování, četnosti založené na statistickém modelování údajů získaných z placebem kontrolovaných klinických studií.
6 Krátce po zahájení léčby ambrisentanem byly hlášeny případy zhoršení dyspnoe nejasné etiologie.
7 Incidence nazální kongesce byla v průběhu léčby ambrisentanem závislá na dávce.
8 Během léčby ambrisentanem byly hlášeny případy autoimunitní hepatitidy, včetně případů zhoršení autoimunitní hepatitidy a poškození jater.
9 Vyrážka včetně erythematózní vyrážky, generalizované vyrážky, papulární vyrážky a svědivé vyrážky.
Popis vybraných nežádoucích účinků Snížení hodnot hemoglobinu
V období po uvedení přípravku na trh byly hlášeny případy anémie, které vyžadovaly podání krevní transfuze (viz bod 4.4). Snížení hodnot hemoglobinu (anémie) bylo častější při užívání ambrisentanu v dávce 10 mg. Ve 12týdenních placebem kontrolovaných klinických studiích fáze 3 došlo ve skupině pacientů užívající ambrisentan ke snížení průměrných koncentrací hemoglobinu a toto snížení bylo zaznamenáno již ve 4. týdnu léčby (snížení o 0,83 g/dl). Průměrné změny oproti výchozímu stavu se stabilizovaly v průběhu následujících 8 týdnů. Celkem u 17 pacientů (6,5 %) ve skupině léčené ambrisentanem došlo ke snížení hemoglobinu o >15 % ve srovnání s výchozími hodnotami, což byl pokles pod dolní hranici normálních hodnot.
Hlášení podezření na nežádoucí účinek
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
U pacientů s PAH léčených ambrisentanem nej sou žádné zkušenosti s podáváním dávek vyšších než
10 mg/den. U zdravých dobrovolníků bylo užití jednorázové dávky 50 mg a 100 mg (5 - 10násobek maximální doporučné dávky) doprovázeno bolestí hlavy, návaly horka/zrudnutím, závratí, nauzeou a zduřením nosní sliznice.
Vzhledem k mechanismu účinku by předávkování ambrisentanem mohlo vést k hypotenzi (viz bod 5.3). Při výrazné hypotenzi může být nezbytná aktivní kardiovaskulární podpora. Specifické antidotum není známo.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihypertenziva, jiná antihypertenziva, ATC kód: C02KX02 Mechanismus účinku
Ambrisentan je perorálně účinný antagonista receptorů pro endotelin (ERA) s afinitou k endotelinovému receptoru typu A (ETA) patřící do třídy kyseliny propionové. Endotelin hraje významnou roli v patofyziologii PAH.
• Ambrisentan je silný/ účinný (Ki 0,016 nM) a vysoce selektivní antagonista ETA (přibližně 4000x selektivnější pro receptory typu ETA v porovnání pro ETB).
• Ambrisentan blokuje subtyp endotelinového receptoru typu ETA, který se nachází zejména v hladké svalovině cév a v srdečních myocytech. Tím zabraňuje endotelinem zprostředkované aktivaci systémů druhého posla, která vede k vazokonstrikci a proliferaci buněk hladkého svalstva.
• V důsledku vyšší afinity ambrisentanu k receptorům ETA než k ETB se předpokládá zachování tvorby vazodilatačně působícího oxidu dusnatého a prostacyklinu, která je zprostředkována receptory ETB.
Klinická účinnost a bezpečnost
Byly provedeny dvě randomizované, dvojitě zaslepené, multicentrické, placebem kontrolované pivotní studie fáze III (ARIES-1 a 2). Ve studii ARIES-1, které se účastnilo 201 pacientů, byl porovnáván ambrisentan v dávce 10 mg a 5 mg s placebem. Ve studii ARIES-2, které se účastnilo 192 pacientů, byl porovnáván ambrisentan v dávce 2,5 mg a 5 mg s placebem. V obou studiích byl ambrisentan přidáván k podpůrné/původní léčbě, která mohla zahrnovat kombinaci digoxinu, antikoagulancií, diuretik, kyslíku a vazodilatancií (blokátory kalciového kanálu, ACE-inhibitory). Pacienti účastnící se studií měli IPAH nebo PAH spojenou s onemocněním pojivové tkáně (PAH-CTD). Většina pacientů měla příznaky II. stupně (38,4 %) nebo III. stupně (55,0 %) WHO funkční klasifikace. Pacienti s preexistujícím jaterním onemocněním (cirhóza nebo klinicky významně zvýšené hodnoty aminotransferáz) a pacienti užívající jinou cílenou léčbu PAH (např. prostanoidy) byli ze studie vyloučeni. Hemodynamické parametry nebyly v těchto studiích hodnoceny.
Primárním cílovým parametrem účinnosti definovaným pro fázi III klinických studií bylo zlepšení zátěžové kapacity, které bylo hodnoceno jako změna vzdálenosti 6 minutové chůze (6MWD) měřené ve 12. týdnu oproti stavu před léčbou. V obou studiích vedla léčba ambrisentanem ve všech podávaných dávkách k významnému zlepšení 6MWD.
Zlepšení průměrné 6MWD upravené k placebovému účinku při užívání dávky 5 mg činilo ve 12. týdnu ve srovnání s výchozí hodnotou 30,6 m (95% IS:2,9 až 58,3; p=0,008) ve studii ARIES-1 a 59,4 m (95% IS: 29,6 až 89,3; p<0,001) ve studii ARIES-2. Zlepšení průměrné 6MWD upravené k placebovému účinku při užívání dávky 10 mg činilo ve studii ARIES-1 51,4 m (95% IS: 26,6 až 76,2; p <0,001).
Byla též provedena předem specifikovaná kombinovaná analýza výsledků studií fáze III (ARIES-C). Zlepšení průměrné 6MWD upravené k placebovému účinku činilo při užívání dávky 5 mg 44,6 m (95 % IS: 24,3 až 64,9; p<0,001) a při užívání dávky 10 mg 52,5 m (95% IS: 28,8 až 76,2; p<0,001).
Ve studii ARIES-2 ambrisentan (ve skupině s kombinovanou dávkou) významně prodloužil dobu do klinického zhoršení PAH ve srovnání s placebem (p<0,001), došlo k 80% snížení rizika výskytu (95% IS: 47% až 92%) sledovaných faktorů. Hodnocení zahrnovalo: úmrtí, transplantaci plic, hospitalizaci pro PAH, atriální septostomii, přidání další medikace k léčbě PAH a kritéria pro předčasné ukončení léčby. Ve skupině s kombinovanou dávkou bylo ve srovnání s placebem pozorováno statisticky významné zlepšení (3,41 ± 6,96) fyzických funkcí hodnocených podle stupnice "SF-36 Health Survey" (-0,20 ± 8,14; p=0,005). Léčba ambrisentanem vedla ke statisticky významnému zlepšení indexu BDI ("Borg Dyspnoe Index") ve 12. týdnu (BDI upravený k placebovému účinku -1,1 (95% IS: -1,8 až -0,4; p=0,019; skupina s kombinovanou dávkou)).
Údaje z dlouhodobých klinických studií
Pacienti, kteří se účastnili studií ARIES 1 a 2, byli způsobilí účastnit se dlouhodobé otevřené rozšířené studie ARIES-E (n=383). Kombinovaná střední expozice léčivému přípravku byla přibližně 145 ± 80 týdnů a maximální expozice byla přibližně 295 týdnů. Hlavním primárním cílovým parametrem studie byla incidence a závažnost nežádoucích účinků souvisejících s dlouhodobou expozicí ambrisentanu, včetně sérových jaterních testů. Nálezy týkající se bezpečnosti pozorované při dlouhodobé expozici ambrisentanu v této studii byly obecně shodné s nálezy pozorovanými ve 12týdenních placebem kontrolovaných studiích.
Pozorovaná pravděpodobnost přežívání u subjektů léčených ambrisentanem (skupina s kombinovanou dávkou ambrisentanu) byla 93% po 1 roce léčby, 85% po 2 letech a 79% po 3 letech léčby.
V otevřené studii (AMB222) byl ambrisentan hodnocen u 36 pacientů a cílem studie bylo posouzení incidence zvýšených koncentrací aminotransferáz v séru u pacientů, u kterých došlo k předchozímu přerušení léčby jinými antagonisty receptorů pro endotelin z důvodu zvýšení koncentrací aminotransferáz. Během průměrné doby 53 týdnů léčby ambrisentanem nebylo u žádného z pacientů zaznamenáno zvýšení sérových koncentrací ALT >3x ULN, které by vyžadovalo trvalé ukončení léčby. U padesáti procent pacientů došlo během této doby ke zvýšení dávky ambrisentanu z 5 mg na 10 mg.
Kumulativní incidence zvýšených sérových koncentrací aminotransferáz >3x ULN ve všech studiích fáze 2 a 3 (včetně příslušné otevřené rozšířené studie) činila 17 subjektů ze 483 při průměrné délce expozice 79,5 týdnů. To odpovídá četnosti 2,3 příhody na 100 pacientoroků expozice ambrisentanu.
V otevřené dlouhodobé rozšířené studii ARIES-E bylo u pacientů léčených ambrisentanem dvouleté riziko rozvoje elevací sérových aminotransferáz >3x ULN 3,9%.
Další klinické údaje
Ve studii fáze 2 (AMB220) bylo po 12 týdnech léčby u pacientů s PAH (n=29) zaznamenáno zlepšení hemodynamických parametrů. Léčba ambrisentanem vedla ke zvýšení průměrného srdečního indexu, ke snížení průměrného tlaku v plicnici a ke snížení průměrné plicní cévní rezistence.
Při léčbě ambrisentanem byl hlášen pokles systolického a diastolického krevního tlaku. V placebem kontrolovaných klinických studiích trvajících 12 týdnů byl průměrný pokles systolického krevního tlaku od výchozích hodnot do ukončení léčby 3 mm Hg a diastolického krevního tlaku 4,2 mm Hg. Průměrný pokles systolického i diastolického krevního tlaku přetrvával po dobu až 4 let během léčby ambrisentanem v dlouhodobé otevřené studii ARIES-E.
V lékové interakční studii u zdravých dobrovolníků nebyl zaznamenán klinicky významný vliv na farmakokinetiku ambrisentanu nebo sildenafilu a tato kombinace byla dobře snášena. Počet pacientů, kteří užívali ambrisentan současně se sildenafilem, činil ve studii ARIES-E 22 pacientů (5,7 %) a ve studii AMB222 17 pacientů (47 %). Žádné další problémy vztahující se k bezpečnosti nebyly u těchto pacientů zaznamenány.
Klinická účinnost v kombinaci s tadalafilem
Multicentrická, dvojitě zaslepená, událostí řízená studie 3. fáze s aktivním komparátorem (AMB112565/AMBITION) byla provedena pro posouzení účinnosti počátečního kombinace ambrisentanu a tadalafilu versus monoterapie buď ambrisentanem nebo tadalafilem samotným na 500 dosud neléčených pacientech s PAH, randomizovaných v poměru 2:1: 1. Žádný z pacientů nedostával pouze placebo. Primární analýza hodnotila skupinu s kombinací oproti poolovaným datům skupin s monoterapií. Podpůrné srovnání skupiny s kombinovanou léčbou versus jednotlivé skupiny s monoterapií bylo též provedeno. Pacienti s výraznou anémií, retencí tekutin nebo vzácnými chorobami sítnice byli vyloučeni v souladu s kritérii investigátorů. Pacienti s hodnotami ALT a AST > dvojnásobku horní hranice normy byli rovněž vyloučeni.
Při vstupu do studie, 96 % pacientů bylo naivních s ohledem na jakoukoli předchozí PAH specifickou léčbu a střední doba od diagnózy do vstupu do studie byla 22 dní. Pacienti, kteří začínali s ambrisentanem 5 mg a tadalafilem 20 mg byli titrováni na 40 mg tadalafilu ve 4. týdnu a 10 mg ambrisentanu v 8. týdnu, pokud nebyly problémy se snášenlivostí. Střední doba dvojitě zaslepené léčby pro kombinovanou terapii byla vyšší než 1,5 roku.
Primárním cílovým parametrem byl čas do prvního výskytu klinického selhání, definovaného jako:
- úmrtí, nebo
- hospitalizace kvůli zhoršení PAH,
- progrese onemocnění;
- dlouhodobá nevyhovující klinická odpověď.
Střední věk všech pacientů byl 54 let (SD 15, rozpětí 18-75 let). Pacienti byli na začátku studie ve II. (31 %) a III. (69 %) funkční třídě dle klasifikace WHO. Idiopatická nebo dědičná PAH byla nejčastější etiologií ve studované populaci (56 %), následovaná PAH v důsledku poškození pojivové tkáně (37 %), PAH související s léčivy a toxiny (3 %), korigované nekomplikované vrozené srdeční vady (2 % ) a HIV (2 %). Pacienti s PAH II. a III. třídy dle klasifikace WHO měli střední výchozí 6MWD 353 metrů.
Výsledky hodnocení
Léčba kombinovanou terapií měla za následek 50% snížení rizika (poměr rizika [PR] 0,502; 95% CI: 0,384 až 0,724; p = 0,0002) složeného parametru účinnosti klinického selhání až do poslední návštěvy ve srovnání s monoterapií [Obrázek 1 a Tabulka 1]. Efekt léčby spočívající v 63% snížení nutnosti hospitalizace při kombinované terapii byl zaznamenán brzy a byl zachován během léčby. Účinnost kombinované terapie vzhledem k primárnímu cíli byla konzistentní ve srovnání s individuální monoterapií a napříč podskupinami podle věku, etnického původu, zeměpisné oblasti, etiologie (IPAH / hPAH a PAH-CTD). Účinek byl významný pro pacienty jak II. tak i III. funkční třídy dle klasifikace
WHO. Obrázek 1
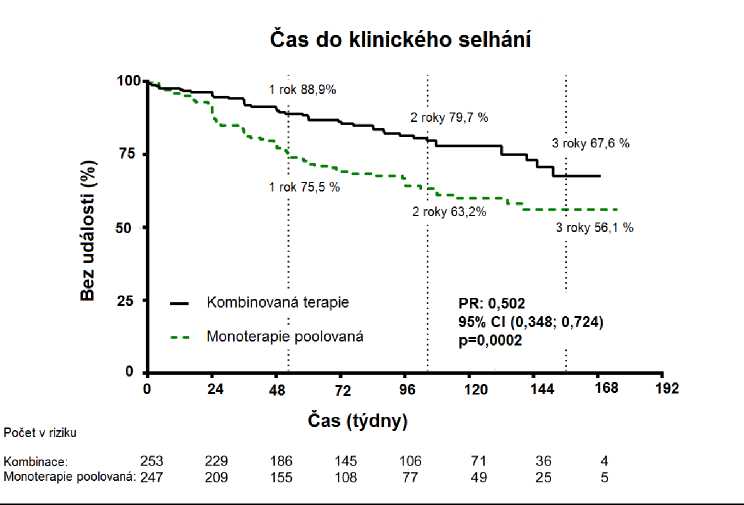
Tabulka 1
|
Ambrisentan + Tadalafil (N=253) |
Monoterapie Poolovaná data (N=247) |
Ambrisentan monoterapie (N=126) |
Tadalafil monoterapie (N=121) | |
|
Čas do události -prvního klinického selhání (určeno) | ||||
|
Klinické selhání, počet (%) |
46 (18%) |
77 (31%) |
43 (34) |
34 (28) |
|
Poměr rizika (95% CI) |
0,502 (0,348 -0,724) |
0,477 (0,314 - 0,723) |
0,528 (0,338 - 0,827) | |
|
Hodnota p, Log-rank test |
0,0002 |
0,0004 |
0,0045 | |
|
Jednotlivé události stanovené jako událost prvního klinického selhání (určeno) | ||||
|
Úmrtí (všechny příčiny) |
9 (4%) |
8 (3%) |
2 (2) |
6 (5) |
|
Hospitalizace pro zhoršení PAH |
10 (4%) |
30 (12%) |
18 (14) |
12 (10) |
|
Progrese nemoci |
10 (4%) |
16 (6%) |
12 (10) |
4 (3) |
|
Dlouhodobě neuspokojivá klinická odpověď |
17 (7%) |
23 (9%) |
11 (9) |
12 (10) |
|
Čas do první hospitalizace pro zhoršení PAH (určeno) | ||||
|
První hospitalizace, časový údaj (%) |
19 (8%) |
44 (18%) |
27 (21%) |
17 (14%) |
|
Poměr rizika (95% CI) |
0.372 |
0.323 |
0.442 | |
|
Hodnota p, Log-rank test |
0.0002 |
<0.0001 |
0.0124 | |
Sekundární_parametry účinnosti
Jako sekundární parametry účinnosti byly testovány:
Tabulka 2
|
Sekundární parametry účinnosti (změna od zahájení studie do 24 týdne) |
Ambrisentan + Tadalafil |
Monoterapie- Poolovaná data |
Rozdíl a interval spolehlivosti |
hodnota p |
|
NT-proBNP (% snížení) |
-67,2 |
-50,4 |
% rozdíl -33,8; 95% CI: -44,8; -20,7 |
p<0,0001 |
|
% subjektů dosahujících uspokojivé klinické odpovědi ve 24. týdnu |
39 |
29 |
Poměr pravděpodobnosti 1,56; 95% CI: 1,05;2,32 |
p=0,026 |
|
6MWD (metry, střední změna) |
49,0 |
23,8 |
22,75m; 95% CI: 12,00; 33,50 |
p<0,0001 |
Idiopatická plicní fibróza
Byla provedena studie hodnotící 492 pacientů (ambrisentan n = 329, placebo n = 163) s idiopatickou plicní fibrózou (IPF), z nichž 11 % mělo sekundární plicní hypertenzi (WHO skupina 3). Studie ale byla ukončena předčasně, když bylo zjištěno, že primární cílový parametr účinnosti nemůže být splněn (studie ARTEMIS-IPF). Ve skupině s ambrisentanem bylo pozorováno 90 případů (27 %) progrese IPF (včetně hospitalizace z důvodu zhoršení respirace) nebo úmrtí ve srovnání s 28 případy (17 %) ve skupině s placebem. Ambrisentan je proto kontraindikován u pacientů s IPF se sekundární plicní hypertenzí nebo bez ní (viz bod 4.3).
5.2 Farmakokinetické vlastnosti
Absorpce
Ambrisentan se u lidí rychle vstřebává. Po perorálním podání je maximálních plazmatických koncentrací ambrisentanu (Cmax) dosaženo přibližně za 1,5 hodiny po užití dávky nalačno i po jídle. Cmax a plocha pod křivkou plazmatických koncentrací (AUC) se v léčebném dávkovém rozmezí zvyšují úměrně s dávkou. Ustáleného stavu je obvykle dosaženo za 4 dny při opakovaném podávání.
U zdravých dobrovolníků, kteří užívali ambrisentan nalačno nebo s jídlem s vysokým obsahem tuků, byla provedena studie hodnotící možný vliv potravy. V této studii došlo ke snížení Cmax o 12 % zatímco AUC zůstala nezměněna. Toto snížení maximální plazmatické koncentrace není klinicky významné a ambrisentan proto lze užívat jak nalačno, tak s jídlem.
Distribuce v organismu
Ambrisentan se ve vysoké míře váže na plazmatické proteiny. In vitro činila vazba ambrisentanu na plazmatické proteiny v průměru 98,8 % a v rozmezí 0,2 - 20 mikrogramů/ml byla nezávislá na koncentraci. Ambrisentan je vázán zejména na albumin (96,5 %) a v menší míře na alfa1-kyselý glykoprotein.
Distribuce ambrisentanu do erytrocytů je nízká, průměrný poměr krev:plazma činí 0,57 u mužů a 0,61 u žen.
Biotransformace
Ambrisentan je nesulfonamidový ERA (kyselina propionová).
Ambrisentan podléhá glukuronidaci pomocí izoenzymů UGT (UGT1A9S, UGT2B7S a UGT1A3S) za vzniku ambrisentan-glukuronidu (13 %). Ambrisentan je také přeměňován cestou oxidativního metabolismu zejména působením CYP3A4 a v menší míře CYP3A5 a CYP2C19 za vzniku 4-hydroxymethyl-ambrisentanu (21 %), který je dále glukuronidován na 4-hydroxymethyl-ambrisentan glukuronid (5 %). Vazebná afinita 4-hydroxymethyl-ambrisentanu k lidskému receptoru pro endotelin je 65x nižší než u ambrisentanu. Nepředpokládá se tedy, že by 4-hydroxymethyl-ambrisentan v koncentracích zaznamenaných v plazmě (přibližně 4 % koncentrace mateřské látky) přispíval k farmakologické aktivitě ambrisentanu.
Údaje in vitro ukazují, že ambrisentan při koncentraci 300 pM vedl k méně než 50% inhibici UGT1A1, UGT1A6, UGT1A9, UGT2B7 (až do 30 %) nebo izoenzymů 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 a 3A4 cytochromu P450 (až 25 %). In vitro, ambrisentan v klinicky relevantních koncentracích nemá inhibiční účinek na lidské transportéry, včetně Pgp, BCRP, MRP2, BSEP, OATP1B1, OATP1B3 a NTCP. Navíc, ambrisentan neindukuje expresi proteinů MRP2, Pgp nebo BSEP v potkaních hepatocytech. Celkově vzato, in vitro data naznačují, že nelze předpokládat, že by ambrisentan v klinicky relevantních koncentracích (Cmax do 3,2 pM v plazmě) ovlivňoval UGT1A1, UGT1A6, UGT1A9, UGT2B7 nebo izoenzymy 1A2, 2A6, 2B6, 2C8, 2C9 , 2C19, 2D6, 2E1, 3A4 cytochromu P450 nebo transport přes BSEP, BCRP, PGP, MRP2, OATP1B1 / 3, nebo NTCP.
Vliv ambrisentanu v ustáleném stavu (10 mg jedenkrát denně) na farmakokinetiku a farmakodynamiku jednorázově podaného warfarinu (25 mg) byl hodnocen pomocí PT a INR u 20 zdravých dobrovolníků. Ambrisentan neměl klinicky významný účinek na farmakokinetiku nebo farmakodynamiku warfarinu. Podobně současné podávání warfarinu neovlivňovalo farmakokinetiku ambrisentanu (viz bod 4.5).
U 19 zdravých dobrovolníků byl hodnocen vliv podávání sildenafilu po dobu 7 dnů (20 mg třikrát denně) na farmakokinetiku ambrisentanu při jednorázovém podání a vliv podávání ambrisentanu po dobu 7 dnů (10 mg jedenkrát denně) na farmakokinetiku sildenafilu při jednorázovém podání. Kromě 13% zvýšení Cmax sildenafilu nedošlo při současném podání s ambrisentanem k žádným jiným změnám farmakokinetických parametrů sildenafilu, N-desmethyl-sildenafilu ani ambrisentanu. Toto mírné zvýšení Cmax sildenafilu není považováno za klinicky významné (viz bod 4.5).
U 23 zdravých dobrovolníků byl hodnocen vliv ambrisentanu v ustáleném stavu (10 mg jedenkrát denně) na farmakokinetiku tadalafilu po jednorázovém podání a vliv tadalafilu v ustáleném stavu (40 mg jedenkrát denně) na farmakokinetiku ambrisentanu po jednorázovém podání. Ambrisentan neměl klinicky významný účinek na farmakokinetiku tadalafilu. Podobně současné podávání s tadalafilem neovlivnilo farmakokinetiku ambrisentanu (viz bod 4.5).
U 16 zdravých dobrovolníků byl hodnocen vliv ketokonazolu při opakovaném podávání (400 mg jedenkrát denně) na farmakokinetiku ambrisentanu při jednorázovém podání 10 mg. Expozice ambrisentanu měřené pomocí AUC(0_infl a Cmax byly zvýšeny o 35 %, respektive o 20 %. Je nepravděpodobné, že by tyto změny v expozici měly klinický význam, a proto může být ambrisentan podáván současně s ketokonazolem.
Účinky opakovaných dávek cyklosporinu A (100 - 150 mg dvakrát denně) na farmakokinetiku ambrisentanu v rovnovážném stavu (5 mg jednou denně) a účinky opakovaných dávek ambrisentanu (5 mg jednou denně) na farmakokinetiku cyklosporinu A v rovnovážném stavu (100 - 150 mg dvakrát denně) byly studovány na zdravých dobrovolnících. Cmax a AUC(0-t) ambrisentanu při mnohočetných dávkách cyklosporinu A stoupalo (48 % resp. 121 %). Na základě těchto změn by dávka ambrisentanu měla být omezena na 5 mg jednou denně, pokud je podáván společně s cyklosporinem A (viz bod 4.2). Mnohočetné dávky ambrisentanu nemají na expozici cyklosporinu A žádný vliv, a proto žádná úprava dávky cyklosporinu A není nutná.
U zdravých dobrovolníků byly studovány účinky akutních a opakovaných dávek rifampicinu (600 mg jedenkrát denně) na farmakokinetiku ambrisentanu (10 mg jedenkrát denně) v ustáleném stavu. Po počátečních dávkách rifampicinu bylo pozorováno přechodné zvýšení hodnoty AUC(0-T) ambrisentanu (121% po první dávce rifampicinu a 116% po druhé dávce rifampicinu), pravděpodobně v důsledku inhibice OATP zprostředkované rifampicinem. Avšak, po podání vícenásobných dávek rifampicinu nebyl do osmého dne pozorován žádný klinicky významný vliv na expozici ambrisentanu. Pacienti, kterým je podáván ambrisentan, musí být po zahájení léčby rifampicinem pečlivě sledováni (viz bod 4.5 a 4.5).
U 15 zdravých dobrovolníků byl hodnocen vliv ambrisentanu (10 mg) při opakovaném podávání na farmakokinetiku digoxinu při jednorázovém podání. Opakované podání ambrisentanu vedlo k mírnému zvýšení AUC0-last a nejnižších koncentrací digoxinu a k 29% zvýšení Cmax digoxinu. Zvýšení expozice digoxinu pozorované při současném podávání opakovaných dávek ambrisentanu nebylo považováno za klinicky významné a úprava dávkování digoxinu tedy není nutná (viz bod 4.5).
U zdravých dobrovolnic byl hodnocen vliv 12denního podávání ambrisentanu (10 mg jedenkrát denně) na farmakokinetiku perorálního antikoncepčního přípravku obsahujícího ethinylestradiol (35pg) a norethisteron (1 mg) po jednorázovém podání. Cmax a AUC(0-X,) ethinylestradiolu byly mírně sníženy (o 8 %, resp. o 4 %) a Cmax a AUC(0-<X)) norethisteronu byly mírně zvýšeny (o 13 %, resp. o 14 %). Tyto změny expozice ethinylestradiolu nebo norethisteronu byly malé a je nepravděpodobné, že by mohly být klinicky významné (viz bod 4.5).
Eliminace z organismu
Ambrisentan a jeho metabolity jsou po hepatální a/nebo extrahepatální metabolizaci vylučovány zejména žlučí. Po perorálním podání se přibližně 22 % podané dávky vyloučí močí; 3,3 % ve formě nezměněného ambrisentanu. Plazmatický eliminační poločas je u lidí v rozmezí 13,6 až 16,5 hodiny.
Zvláštní skupiny pacientů
Výsledky populační farmakokinetické analýzy u zdravých dobrovolníků a pacientů s PAH prokázaly, že farmakokinetika ambrisentanu není významně ovlivněna pohlavím ani věkem (viz bod 4.2).
Porucha funkce ledvin
Ambrisentan nepodléhá významnému renálnímu metabolismu ani renální clearance (vylučování). V populační farmakokinetické analýze byla clearance kreatininu statisticky významnou veličinou ovlivňující clearance ambrisentanu po perorálním podání. Snížení clearance po perorálním podání je u pacientů se středně závažným renálním poškozením mírné (20-40 %) a je tedy nepravděpodobné, že by mohlo být klinicky významné. U pacientů se závažným poškozením ledvin je však nutná opatrnost (viz bod 4.2).
Porucha funkce jater
Hlavní metabolickou cestou ambrisentanu je glukuronidace a oxidace s následným vylučováním žlučí a mohlo by se tedy předpokládat, že jaterní poškození může zvýšit expozici (Cmax a AUC) ambrisentanu. V populační farmakokinetické analýze se clearance po perorálním podání snižovala v závislosti na zvyšujících se hladinách bilirubinu. Vliv bilirubinu je však mírný (ve srovnání s pacientem s běžnou hodnotou bilirubinu 0,6 mg/dl by měl pacient s hladinou bilirubinu zvýšenou na 4,5 mg/dl přibližně o 30 % nižší clearance ambrisentanu po perorálním podání). Farmakokinetika ambrisentanu nebyla u pacientů s jaterním poškozením (s cirhózou nebo bez cirhózy) hodnocena. Léčba ambrisentanem se proto nemá zahajovat u pacientů s poruchou funkce jater nebo s klinicky významně zvýšenými hodnotami jaterních aminotransferáz (>3x ULN) (viz body 4.3 and 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Vzhledem k hlavnímu farmakologickému účinku této třídy léčiv by mohlo vést podání vysoké jednorázové dávky ambrisentanu (tj. předávkování) ke snížení krevního tlaku a k možné hypotenzi a příznakům souvisejícím s vazodilatací.
Nebylo prokázáno, že by ambrisentan inhiboval transport žlučových kyselin nebo že by byl významně hepatotoxický.
Po dlouhodobém podávání byly u hlodavců v expozicích nižších, než odpovídají terapeutickým hladinám u člověka pozorovány zánět a změny epitelu nosní dutiny. U psů byla při dlouhodobém podávání vysokých dávek ambrisentanu v expozicích vyšších než 20násobek expozice u člověka pozorována mírná zánětlivá odpověď.
U potkanů, kterým byl podáván ambrisentan v expozicích 3krát vyšších než j sou klinické hodnoty AUC, byla v nosní dutině zaznamenána hyperplazie nosní kosti (kosti čichové). Hyperplazie nosní kosti nebyla při podávání ambrisentanu pozorována u myší a psů. Na základě zkušeností s dalšími sloučeninami je hyperplazie nosní skořepy u potkanů zjevnou odpovědí na nosní zánět.
Při testování in vitro na buňkách savců byl ambrisentan ve vysokých koncentracích klastogenní. Mutagenní ani genotoxický účinek ambrisentanu nebyl v testech na bakteriích ani ve dvou in vivo studiích na hlodavcích prokázán.
Ve 2letých studiích perorálního podávání u potkanů a myší nebyl prokázán karcinogenní potenciál. Byl zaznamenán malý nárůst incidence fibroadenomů prsu (benigních tumorů) a to pouze u samců potkanů při nejvyšší dávce. Systémová expozice ambrisentanu byla u samců potkanů při této dávce (na základě AUC v ustáleném stavu) 6násobná ve srovnání s expozicí, které je dosahováno při klinické dávce 10 mg/den.
Ve studiích toxicity při opakovaném perorálním podávání a ve studiích fertility provedených na potkaních a myších samcích mimo bezpečnostní rozpětí byla zaznamenána testikulární tubulární atrofie, která byla v některých případech spojena s aspermií. V průběhu hodnoceného období bez podávání dávek se testikulární změny zcela neupravily. Nicméně ve studiích na psech, trvajících až 39 týdnů při expozici 35krát vyšší než je expozice u lidí určená na základě AUC, nebyly pozorovány žádné testikulární změny. Při žádné z testovaných dávek (až 300 mg/kg/den) nebyly u samců potkanů pozorovány žádné účinky ambrisentanu na motilitu spermií. Při dávkách 300 mg/kg/den bylo pozorováno mírné (<10 %) snížení procenta morfologicky normálních spermií; při dávkách 100 mg/kg/den (> 9násobek klinické expozice 10 mg/den) však toto snížení pozorováno nebylo. Vliv ambrisentanu na fertilitu u mužů není znám.
Ambrisentan měl u potkanů a králíků teratogenní účinky. Anomálie dolní čelisti, jazyka a/nebo patra byly pozorovány ve všech testovaných dávkách. Ve studiích na potkanech byl navíc zaznamenán zvýšený výskyt defektů komorového septa, defektů kmenových cév, anomálií štítné žlázy a thymu, osifikace basisphenoidální kosti a lokalizace umbilikální arterie na levé straně močového měchýře místo na pravé straně.
Teratogenita je očekávaným nežádoucím účinkem celé třídy antagonistů receptorů pro endotelin.
Při expozicích 3krát převyšujících AUC maximálních doporučených dávek u lidí, vedlo podávání ambrisentanu potkaním samicím v období od pozdní březosti po laktaci k nežádoucímu ovlivnění mateřského chování, ke sníženému přežívání mláďat a k poškození reprodukční schopnosti potomstva (v pitevním nálezu byla pozorována malá varlata).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety Monohydrát laktosy Mikrokrystalická celulosa Sodná sůl kroskarmelosy Magnesium-stearát
Potah tablety
Částečně hydrolyzovaný polyvinylalkohol Mastek (E553b)
Oxid titaničitý (E171)
Makrogol/PEG 3350 Sójový lecithin (E322)
Hlinitý lak červeně Allura AC (E129)
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
PVC/PVDC/Al blistry. Velikost balení s jednodávkovými blistry po 10x1 nebo 30x1 potahovaných tabletách. Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd
980 Great West Road
Brentford
Middlesex
TW8 9GS
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/451/001
EU/1/08/451/002
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. dubna 2008 Datum posledního prodloužení: 24.dubna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky na adrese http://www.ema.europa.eu.
NÁZEV PŘÍPRAVKU
1.
Volibris 10 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna tableta obsahuje ambrisentanum 10 mg.
Pomocné látky se známým účinkem:
Jedna tableta obsahuje přibližně 90 mg laktosy (ve formě monohydrátu), přibližně 0,25 mg sójového lecithinu (E322) a přibližně 0,45 mg hlinitého laku červeně Allura AC (E129).
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta).
Tmavě růžové, oválné, vypouklé potahované tablety s vyraženým "GS" na jedné straně a "KE3" na straně druhé.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Volibris je indikován k léčbě plicní arteriální hypertenze (PAH) II. až III. funkční třídy (FC) dle klasifikace WHO u dospělých pacientů včetně použití v kombinované terapii (viz bod 5.1). Účinnost byla prokázána u idiopatické PAH (IPAH) a PAH spojené s onemocněním pojivové tkáně.
4.2 Dávkování a způsob podání
Léčba musí být zahájena lékařem, který má zkušenosti s léčbou PAH.
Dávkování
Ambrisentan v monoterapii
Volibris se užívá perorálně v počáteční dávce 5 mg jedenkrát denně a dávka může být zvýšena na 10 mg denně v závislosti na klinické odpovědi a snášenlivosti.
Ambrisentan v kombinaci s tadalafilem
Při užívání v kombinaci s tadalafilem má být Volibris titrován do dávky 10 mg jednou denně.
Ve studii AMBITION pacienti užívali 5 mg ambrisentanu denně po dobu prvních 8 týdnů před zvýšením až na 10 mg, v závislosti na snášenlivosti (viz bod 5.1). Při užívání v kombinaci s tadalafilem, pacienti začínali s 5 mg ambrisentanu a 20 mg tadalafilu. V závislosti na snášenlivosti byla dávka tadalafilu zvýšena po 4 týdnech na 40 mg a dávka ambrisentanu byla zvýšena po 8 týdnech na 10 mg. Tohoto dosáhlo více než 90 % pacientů. Dávky také mohly být sníženy v závislosti na snášenlivosti.
Na základě omezených údajů lze usuzovat, že náhlé přerušení léčby ambrisentanem není spojeno s opětovným zhoršením PAH.
Pokud je ambrisentan podáván společně s cyklosporinem A, měla by být jeho dávka omezena na 5 mg jednou denně a pacient by měl být pečlivě monitorován (viz bod 4.5 a 5.2).
Zvláštní populace
Starší _ pacienti
U pacientů starších 65 let není nutná žádná úprava dávkování (viz bod 5.2).
Pacienti s _poruchou renální _funkce
U pacientů s poruchou renální funkce není nutná žádná úprava dávkování (viz bod 5.2). U jedinců s těžkou poruchou renální funkce (clearance kreatininu <30 ml/min) jsou s podáváním ambrisentanu jen omezené zkušenosti; proto u této skupiny pacientů musí být léčba zahajována s opatrností a zvláštní opatrnost je nezbytná zejména při zvýšení dávky ambrisentanu na 10 mg.
Pacienti s _poruchou hepatální _funkce
Ambrisentan nebyl hodnocen u jedinců s poruchou funkce jater (s cirhózou nebo bez cirhózy). Vzhledem k tomu, že hlavní metabolickou cestou ambrisentanu je glukuronidace a oxidace s následným vylučováním žlučí, lze předpokládat, že jaterní poškození může zvýšit expozici (Cmax a AUC) ambrisentanu. Léčba ambrisentanem se nesmí zahajovat u pacientů s těžkým jaterním poškozením nebo s klinicky významně zvýšenými hodnotami jaterních aminotransferáz (více než trojnásobek horního limitu normálních hodnot (>3x ULN); viz body 4.3 and 4.4).
Pediatrická populace
Bezpečnost a účinnost ambrisentanu u dětí a dospívajících mladších 18 let nebyla stanovena. Nejsou dostupné žádné údaje.
Způsob podání
Doporučuje se užívat tablety celé, nalačno nebo s jídlem. Doporučuje se, aby tableta nebyla dělena, drcena ani žvýkána.
4.3 Kontraindikace
• Hypersenzitivita na léčivou látku, sóju nebo kteroukoli pomocnou látku tohoto přípravku uvedenou v bodě 6.1.
• Těhotenství (viz bod 4.6).
• Ženy v reprodukčním věku, které nepoužívají účinnou antikoncepci (viz body 4.4 a 4.6).
• Kojení (viz bod 4.6).
• Těžké jaterní poškození (s cirhózou nebo bez cirhózy) (viz bod 4.2).
• Výchozí hodnoty jaterních aminotransferáz (aspartátaminotransferázy (AST) a/nebo alaninaminotransferázy (ALT) >3x ULN (viz body 4.2 and 4.4).
• Idiopatická plicní fibróza (IPF) se sekundární plicní hypertenzí nebo bez ní (viz bod 5.1).
4.4 Zvláštní upozornění a opatření pro použití
Ambrisentan nebyl hodnocen u dostatečného množství pacientů, aby bylo možno určit poměr prospěchu/rizika u pacientů s PAH I. funkční třídy dle klasifikace WHO.
U pacientů s PAH IV. funkční třídy dle klasifikace WHO nebyla účinnost ambrisentanu v monoterapii hodnocena. Při zhoršení klinického stavu by měla být zvážena léčba, která se doporučuje při těžkém stadiu tohoto onemocnění (např. epoprostenol).
Jaterní funkce
S PAH bývají spojeny poruchy jaterní ch funkcí. Při léčbě ambrisentanem byly zaznamenány případy odpovídající autoimunitní hepatitidě, včetně možné exacerbace již existující autoimunitní hepatitidy, poškození jater a zvýšené hodnoty jaterních enzymů vznikající v možné souvislosti s léčbou (viz body 4.8 a 5.1). Před zahájením léčby ambrisentanem by proto měly být posouzeny jaterní aminotransferázy (ALT a AST) a léčba nesmí být zahájena u pacientů s výchozími hodnotami ALT a/nebo AST >3xULN (viz bod 4.3).
Pacienti by měli být sledováni s ohledem na příznaky poškození jater a doporučuje se monitorovat hodnoty ALT a AST každý měsíc. Pokud dojde u pacienta k rozvoji přetrvávajícího, nejasného, klinicky významného zvýšení hodnot ALT a/nebo AST nebo je-li zvýšení hodnot ALT a/nebo AST spojeno se známkami nebo příznaky jaterního poškození (např. žloutenka), má být terapie ambrisentanem přerušena.
U pacientů bez klinických příznaků jaterního poškození nebo žloutenky může být po úpravě hodnot jaterních enzymů zváženo opětovné zahájení léčby ambrisentanem. Doporučuje se konzultace s hepatologem.
Koncentrace hemoglobinu
Při léčbě antagonisty receptorů pro endotelin (ERA) včetně ambrisentanu byly zaznamenány snížené koncentrace hemoglobinu a hematokritu. K tomuto snížení došlo většinou v průběhu prvních 4 týdnů léčby a později se hodnoty hemoglobinu obvykle stabilizovaly. Průměrné snížení koncentrace hemoglobinu od výchozích hodnot (v rozmezí od 0,9 do 1,2 g/dl) přetrvávalo po dobu až 4 let během léčby ambrisentanem v dlouhodobém otevřeném rozšíření pivotních klinických studiích fáze 3. V období po uvedení přípravku na trh byly hlášeny případy anémie vyžadující krevní transfuzi (viz bod 4.8).
U pacientů s klinicky významnou anémií se zahájení léčby ambrisentanem nedoporučuje. Doporučuje se, aby byly v průběhu léčby ambrisentanem hodnoty hemoglobinu a/nebo hematokritu monitorovány, např. v 1. měsíci, ve 3. měsíci a dále v pravidelných intervalech v souladu s klinickou praxí. Pokud je zaznamenán klinicky významný pokles hemoglobinu nebo hematokritu a jiné příčiny tohoto poklesu jsou vyloučeny, mělo by být zváženo snížení dávky nebo přerušení léčby. Výskyt anémie se zvýšil, když byl ambrisentan podáván v kombinaci s tadalafilem (frekvence výskytu nežádoucích účinků 15 %) v porovnání s výskytem anémie, kdy byly ambrisentan a tadalafil podávány samostatně jako monoterapie (7% a 11%).
Retence tekutin
Při užívání antagonistů receptorů pro endotelin (ERA) včetně ambrisentanu byl zaznamenán periferní edém. Periferní edém zaznamenaný v klinických studiích s ambrisentanem byl ve většině případů mírný až středně závažný, je však možné, že se vyskytoval častěji a že byl závažnější u pacientů >65 let. Periferní edém byl hlášen častěji při užívání dávky 10 mg ambrisentanu v krátkodobých klinických studiích (viz bod 4.8).
Po uvedení přípravku na trh byly hlášeny případy retence tekutin, ke kterým došlo během několika týdnů po zahájení léčby ambrisentanem. V některých případech bylo k obnovení rovnováhy tekutin nebo u dekompenzovaného srdečního selhání nutné podání diuretik nebo hospitalizace. Pokud je u pacientů zaznamenána již existující zvýšená retence tekutin, je třeba ji vhodným způsobem řešit ještě před zahájením léčby ambrisentanem.
Dojde-li během léčby ambrisentanem k rozvoji klinicky významné retence tekutin (s nárůstem tělesné hmotnosti nebo bez něj), je nutno posoudit příčinu tohoto stavu, kterou může být léčba ambrisentanem nebo již existující srdeční selhávání, a je třeba zvážit možnost eventuální specifické léčby nebo přerušení léčby ambrisentanem. Výskyt periferního edému se zvýšil, když byl ambrisentan podáván v kombinaci s tadalafilem (frekvence výskytu nežádoucího účinků 45 %), v porovnání s výskytem periferního edému, kdy byly ambrisentan a tadalafil podávány jako monoterapie (38% a 28%). Výskyt periferního edému byl nejvyšší během prvního měsíce po zahájení léčby.
Ženy v reprodukčním věku
Léčba přípravkem Volibris může být u žen v reprodukčním věku zahájena pouze v případě, že je výsledek jejich těhotenského testu provedeného před zahájením léčby negativní a pouze při používání účinné antikoncepce. V případě pochybností, jakou antikoncepci doporučit konkrétní pacientce, by měla být zvážena konzultace s gynekologem. V průběhu léčby ambrisentanem se doporučuje provádět jednou měsíčně těhotenské testy (viz body 4.3 a 4.6).
Plicní venookluzivní nemoc
U pacientů s plicní venookluzivní nemocí byly při užívání vazodilatačních léčivých přípravků, jako jsou například ERA, hlášeny případy vzniku edému plic. Tudíž dojde-li při léčbě ambrisentanem u pacientů s PAH ke vzniku akutního edému plic, musí být zvažována možnost přítomnosti venookluzivní nemoci.
Současné podávání s jinými léčivými přípravky
Pacienti, kterým je podáván ambrisentan, musí být po zahájení léčby rifampicinem pečlivě sledováni (viz bod 4.5 a 5.2).
Pomocné látky
Tablety přípravku Volibris obsahují laktosu. Tento přípravek by neměli užívat pacienti se vzácnými dědičnými problémy s intolerancí galaktosy, vrozeným nedostatkem laktasy nebo malabsorpcí glukosy a galaktosy.
Tablety přípravku Volibris obsahují azobarvivo hlinitý lak červeně Allura AC (E129), které může vyvolat alergickou reakci.
Tablety přípravku Volibris obsahují lecithin získaný ze sóji. Pokud je pacient přecitlivělý na sóju, nesmí ambrisentan užívat (viz bod 4.3).
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Ambrisentan neinhibuje ani neindukuje enzymy I. fáze nebo II. fáze metabolické přeměny léčiva v klinicky významných koncentracích v předklinických studiích in vitro a in vivo. Z této skutečnosti lze usuzovat na nízký potenciál ambrisentanu k ovlivnění profilu léčivých přípravků metabolizovaných touto cestou.
Možná indukce aktivity CYP3A4 způsobená ambrisentanem byla hodnocena u zdravých dobrovolníků. Výsledky tohoto hodnocení nenasvědčují tomu, že by měl ambrisentan indukční účinek na izoenzym CYP3A4.
Cvklosporin A
Společné podávání ambrisentanu a cyklosporinu A u zdravých dobrovolníků vedlo v rovnovážném stavu ke dvojnásobnému zvýšení expozice ambrisentanu. To může být v důsledku cyklosporinem A způsobené inhibice transportérů a metabolických enzymů zahrnutých do farmakokinetiky ambrisentanu. Proto by dávka ambrisentanu měla být omezena na 5 mg jednou denně, pokud je podáván společně s cyklosporinem A (viz bod 4.2). Mnohočetné dávky ambrisentanu neměly na expozici cyklosporinu A vliv, a proto není úprava dávky cyklosporinu A nutná.
Rifampicin
Současné podávání rifampicinu (inhibitor transportního proteinu pro organické anionty [OATP], silný induktor CYP3A a 2C19 a induktor P-gp a uridindifosfát glukuronosyltransferáz [UGT]) s ambrisentanem bylo spojeno s přechodným (přibližně dvojnásobným) zvýšením expozice ambrisentanu po počátečních dávkách podaných zdravým dobrovolníkům. Do osmého dne se však hladina upravila a podávání rifampicinu v ustáleném stavu nemělo žádný klinicky relevantní vliv na expozici ambrisentanu. Pacienti, kterým je podáván ambrisentan, musí být po zahájení léčby rifampicinem pečlivě sledováni (viz body 4.5 a 5.2).
Inhibitory fosfodiesterázy
Současné podávání ambrisentanu s inhibitory fosfodiesterázy, a to se sildenafilem nebo tadalafilem (obě látky jsou substráty pro CYP3A4) zdravým dobrovolníkům nemělo významný vliv na farmakokinetiku inhibitorů fosfodiesterázy nebo ambrisentanu (viz bod 5.2).
Další cílená léčba PAH
Účinnost a bezpečnost ambrisentanu při současné další léčbě PAH (např. prostanoidy a stimulátory rozpustné guanylátcyklázy) nebyly v kontrolovaných klinických studiích u pacientů s PAH specificky hodnoceny (viz bod 5.1). Žádné specifické lékové interakce se stimulátory rozpustné guanylátcyklázy nebo prostanoidy se neočekávají na základě známých dat o biotransformaci (viz bod 5.2). Nicméně, studie specifických lékových interakcí s těmito léky nebyly provedeny.
Proto se při současném užívání těchto léčiv doporučuje postupovat s opatrností.
Perorální antikoncepce
V klinické studii se zdravými dobrovolníky neovlivnilo podávání ambrisentanu v dávce 10 mg jedenkrát denně v ustáleném stavu významně farmakokinetiku složek kombinované perorální antikoncepce - ethinylestradiolu a norethisteronu - po jednorázovém podání (viz bod 5.2). Na základě této farmakokinetické studie se nepředpokládá, že by ambrisentan významně ovlivňoval expozici antikoncepčním přípravkům s obsahem estrogenu nebo progestinu.
Warfarin
Ve studii se zdravými dobrovolníky neměl ambrisentan žádný vliv na farmakokinetiku warfarinu v ustáleném stavu ani na antikoagulační aktivitu warfarinu (viz bod 5.2). Podobně warfarin neměl klinicky signifikantní vliv na farmakokinetiku ambrisentanu. Navíc u pacientů neměl ambrisentan celkově žádný vliv na týdenní dávku antikoagulancií typu warfarinu, protrombinový čas (PT) ani na mezinárodní normalizovaný poměr (INR).
Ketokonazol
Podávání ketokonazolu (silného inhibitoru CYP3A4) v rovnovážném stavu nevedlo ke klinicky významnému zvýšení expozice ambrisentanu (viz bod 5.2).
Vliv ambrisentanu na xenobiotické transportéry
In vitro ambrisentan při klinicky relevantních koncentracích u lidí neinhibuje transportéry, včetně P-glykoproteinu (Pgp), proteinu resistence karcinomu prsu (BCRP), izoformy 2 proteinu mnohočetné lékové rezistence (MRP2), exportní pumpu žlučových kyselin (BSEP), polypeptidy transportující organické ionty (OATP1B1 and OATP1B3) a na sodíku závislý kotransportér taurocholátu sodného (NTCP).
Ambrisentan je substrátem pro eflux zprostředkovaný Pgp.
In vitro studie prováděné na potkaních hepatocytech taktéž prokázaly, že ambrisentan neindukuje expresi proteinů Pgp, BSEP ani MRP2 .
Podávání ambrisentanu nemělo v ustáleném stavu žádný klinicky významný účinek na farmakokinetiku digoxinu (substrátu Pgp) po jeho jednorázovém podání (viz bod 5.2).
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Léčba ambrisentanem může být u žen ve fertilním věku zahájena pouze v případě, že je výsledek jejich těhotenského testu provedeného před zahájením léčby negativní a pouze při používání účinné antikoncepce. V průběhu léčby přípravkem Volibris se doporučuje provádět jednou měsíčně těhotenské testy.
Ambrisentan je v těhotenství kontraindikován (viz bod 4.3). Ve studiích na zvířatech byla prokázána teratogenita ambrisentanu. S podáváním u lidí nejsou žádné zkušenosti.
Ženy užívající ambrisentan musí být upozorněny na riziko poškození plodu a pokud dojde k otěhotnění, musí být zahájena alternativní léčba (viz body 4.3, 4.4 a 5.3).
Kojení
Není známo, zda je ambrisentan vylučován do lidského mateřského mléka. Vylučování ambrisentanu do mléka nebylo u zvířat hodnoceno. Z tohoto důvodu je u pacientek užívajících ambrisentan kojení kontraindikováno (viz bod 4.3).
Fertilita u mužů
Dlouhodobé podávání antagonistů receptorů pro endotelin (včetně ambrisentanu) u samců laboratorních zvířat bylo spojeno s rozvojem tubulární atrofie varlat (viz bod 5.3). Ačkoli nebyl ve studii ARIES-E nalezen žádný zřejmý důkaz škodlivého vlivu dlouhodobé expozice ambrisentanu na počet spermií, chronické podávání ambrisentanu bylo spojováno se změnami markerů spermatogeneze. Bylo pozorováno snížení plazmatických koncentrací inhibinu B a zvýšení plazmatických koncentrací FSH. Vliv na fertilitu u mužů není znám, ale narušení spermatogeneze nelze vyloučit. V klinických studiích nebylo dlouhodobé užívání ambrisentanu spojeno se změnami plazmatických hladin testosteronu.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Ambrisentan má mírný nebo středně významný vliv na schopnost řídit a obsluhovat stroje. Při zvažování schopnosti pacienta provádět činnosti, které vyžadují rozhodovací, motorické a kognitivní schopnosti je třeba vzít v úvahu klinický stav pacienta a profil nežádoucích účinků ambrisentanu (jako je hypotenze, závrať, asténie a únava) (viz bod 4.8). Pacienti by měli být před řízením a obsluhou strojů upozorněni, jak je může ambrisentan ovlivnit.
4.8 Nežádoucí účinky
Souhrn bezpečnostního profilu
Bezpečnost ambrisentanu jako monoterapie a/nebo v kombinaci byla hodnocena v klinických studiích s více než 1200 pacienty s PAH (viz bod 5.1). Nežádoucí účinky vycházející z údajů 12týdenní placebem kontrolované klinické studie, seřazené podle tříd orgánových systémů a četnosti, jsou zahrnuty níže. Informace z dlouhodobých, placebem nekontrolovaných studií (ARIES-E a AMBITION (kombinace s tadalafilem)), jsou též uvedeny níže. Žádné dříve nepozorované nežádoucí účinky nebyly při dlouhodobé léčbě nebo při kombinaci ambrisentanu s tadalafilem identifikovány. Bezpečnostní profil byl při delším pozorování vycházejícím z nekontrolovaných studií (průměrná doba sledování 79 týdnů) podobný bezpečnostnímu profilu vycházejícímu z krátkodobých studií. Údaje získané z rutinního farmakovigilančního sledování jsou rovněž uvedeny.
Nejčastěji pozorovanými nežádoucími účinky v souvislosti s léčbou ambrisentanem byly periferní edém, retence tekutin a bolest hlavy (zahrnující bolest hlavy způsobenou onemocněním vedlejších nosních dutin a migrénu). Vyšší dávky (10 mg) byly spojovány s vyšší incidencí těchto nežádoucích účinků a periferní edém měl u pacientů starších 65 let závažnější projevy v krátkodobých klinických studiích (viz bod 4.4).
Seznam nežádoucích účinků v tabulce
|
Ambrisentan (ARIES-C a po uvedení na trh) |
Ambrisentan (AMBITION a ARIES-E) |
Kombinace s tadalafilem (AMBITION) | |
|
Poruchy krve a lymfatického systému | |||
|
Anémie (pokles hemoglobinu, pokles hematokritu) |
Časté1 |
Velmi časté |
Velmi časté |
|
Poruchy imunitního systému | |||
|
Reakce přecitlivělosti (např. angioedém, vyrážka, pruritus) |
Méně časté |
Časté |
Časté |
|
Poruchy nervového systému | |||
|
Bolest hlavy (včetně bolesti dutin v hlavě , migréna) |
Velmi časté2 |
Velmi časté |
Velmi časté |
|
Závrať |
Časté3 |
Velmi časté |
Velmi časté |
|
Oční poruchy | |||
|
Rozmazané vidění, poruchy zraku |
Není známo4 |
Časté |
Časté |
|
Poruchy ucha a ušního labyrintu | |||
Skupiny četností jsou definovány následujícím způsobem: velmi časté (>1/10), časté (> 1/100 až <1/10), méně časté (> 1/1000 až <1/100), vzácné (>1/10 000 až <1/1000), velmi vzácné (<1/10 000) a není známo (z dostupných údajů nelze určit). U nežádoucích účinků závislých na dávce je uváděna skupina četností odpovídající vyšší dávce ambrisentanu. Skupiny četností neodrážejí další faktory, jako jsou rozdílná délka studií, zdravotní stav a laboratorní parametry pacienta před léčbou. Skupiny četností nežádoucích účinků určené na základě zkušeností z klinických studií nemusejí odrážet četnost nežádoucích účinků, které se objeví v průběhu běžné klinické praxe. V každé skupině četností jsou nežádoucí účinky seřazeny podle klesající závažnosti._
|
Tinnitus |
NH |
NH |
Časté |
|
Náhlá ztráta sluchu |
NH |
NH |
Méně časté |
|
Srdeční poruchy | |||
|
Srdeční selhání |
Časté5 |
Časté |
Časté |
|
Palpitace |
Časté |
Velmi časté |
Velmi časté |
|
Cévní poruchy | |||
|
Časté3 |
Časté |
Časté | |
|
Návaly horka/zrudnutí |
Časté |
Časté |
Velmi časté |
|
Synkopa |
Méně časté3 |
Časté |
Časté |
|
Respirační, hrudní a mediastinální poruchy | |||
|
Epistaxe |
Časté3 |
Časté |
Časté |
|
Časté3,6 |
Velmi časté |
Velmi časté | |
|
Kongesce horních cest dýchacích (včetně nosu a vedlejších nosních dutin), sinusitida, nazofaryngitida, rinitida |
Časté7 | ||
|
Nazofaryngitida |
Velmi časté |
Velmi časté | |
|
Sinusistida, rinitida |
Časté |
Časté | |
|
Ucpaný nos |
Velmi časté |
Velmi časté | |
|
Gastrointestinální poruchy | |||
|
Časté3 | |||
|
Velmi časté |
Velmi časté | ||
|
Časté |
Velmi časté | ||
|
Velmi časté |
Velmi časté | ||
|
Časté |
Časté |
Časté | |
|
Zácpa |
Časté |
Časté |
Časté |
|
Poruchy jater a žlučových cest | |||
|
Poškození jater (viz bod 4.4) |
Méně časté3,8 |
NH |
NH |
|
Autoimunitní hepatitida (viz bod 4.4) |
Méně časté3,8 |
NH |
NH |
|
Zvýšení jaterních transamináz |
Časté3 |
NH |
NH |
|
Poruchy kůže a podkožní tkáně | |||
|
NH |
Časté9 |
Velmi časté9 | |
|
Celkové a jinde nezařazené poruchy a lokální reakce po podání | |||
|
Periferní otok, retence tekutin |
Velmi časté |
Velmi časté |
Velmi časté |
|
Bolest/nepříjemný pocit na hrudi |
Časté |
Časté |
Velmi časté |
|
Astenie |
Časté3 |
Časté |
Časté |
|
Únava |
Časté3 |
Velmi časté |
Velmi časté |
NH - nebylo hlášeno
1 Viz bod „Popis vybraných nežádoucích účinků".
2 Bolesti hlavy se objevují častěji při užívání ambrisentanu v dávce 10 mg.
3 Údaje získané z rutinního farmakovigilančního sledování a četnosti založené na zkušenostech z placebem kontrolovaných klinických studií.
4 Data odvozená z rutinního farmakovigilančního sledování
5 Většina hlášených případů srdečního selhání byla spojována s retencí tekutin. Údaje odvozené z rutinního farmakovigilančního sledování, četnosti založené na statistickém modelování údajů získaných z placebem kontrolovaných klinických studií.
6 Krátce po zahájení léčby ambrisentanem byly hlášeny případy zhoršení dyspnoe nejasné etiologie.
7 Incidence nazální kongesce byla v průběhu léčby ambrisentanem závislá na dávce.
8 Během léčby ambrisentanem byly hlášeny případy autoimunitní hepatitidy, včetně případů zhoršení autoimunitní hepatitidy a poškození jater.
9 Vyrážka včetně erythematózní vyrážky, generalizované vyrážky, papulární vyrážky a svědivé vyrážky.
Popis vybraných nežádoucích účinků Snížení hodnot hemoglobinu.
V období po uvedení přípravku na trh byly hlášeny případy anémie, které vyžadovaly podání krevní transfuze (viz bod 4.4). Snížení hodnot hemoglobinu (anémie) bylo častější při užívání ambrisentanu v dávce 10 mg. Ve 12týdenních placebem kontrolovaných klinických studiích fáze 3 došlo ve skupině pacientů užívající ambrisentan ke snížení průměrných koncentrací hemoglobinu a toto snížení bylo zaznamenáno již ve 4. týdnu léčby (snížení o 0,83 g/dl). Průměrné změny oproti výchozímu stavu se stabilizovaly v průběhu následujících 8 týdnů. Celkem u 17 pacientů (6,5 %) ve skupině léčené ambrisentanem došlo ke snížení hemoglobinu o >15% ve srovnání s výchozími hodnotami, což byl pokles pod dolní hranici normálních hodnot.
Hlášení podezření na nežádoucí účinek
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
U pacientů s PAH léčených ambrisentanem nej sou žádné zkušenosti s podáváním dávek vyšších než
10 mg/den. U zdravých dobrovolníků bylo užití jednorázové dávky 50 mg a 100 mg (5 - 10násobek maximální doporučné dávky) doprovázeno bolestí hlavy, návaly horka/zrudnutím, závratí, nauzeou a zduřením nosní sliznice.
Vzhledem k mechanismu účinku by předávkování ambrisentanem mohlo vést k hypotenzi (viz bod 5.3). Při výrazné hypotenzi může být nezbytná aktivní kardiovaskulární podpora. Specifické antidotum není známo.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antihypertenziva, jiná antihypertenziva, ATC kód: C02KX02
Mechanismus účinku
Ambrisentan je perorálně účinný antagonista receptorů pro endotelin (ERA) s afinitou k endotelinovému receptoru typu A (ETA) patřící do třídy kyseliny propionové. Endotelin hraje významnou roli v patofyziologii PAH.
• Ambrisentan je silný/ účinný (Ki 0,016 nM) a vysoce selektivní antagonista ETA (přibližně 4000x selektivnější pro receptory typu ETA v porovnání pro ETB).
• Ambrisentan blokuje subtyp endotelinového receptoru typu ETA, který se nachází zejména v hladké svalovině cév a v srdečních myocytech. Tím zabraňuje endotelinem zprostředkované aktivaci systémů druhého posla, která vede k vazokonstrikci a proliferaci buněk hladkého svalstva.
• V důsledku vyšší afinity ambrisentanu k receptorům ETA než k ETB se předpokládá zachování tvorby vazodilatačně působícího oxidu dusnatého a prostacyklinu, která je zprostředkována receptory ETB.
Klinická účinnost a bezpečnost
Byly provedeny dvě randomizované, dvojitě zaslepené, multicentrické, placebem kontrolované pivotní studie fáze III (ARIES-1 a 2). Ve studii ARIES-1, které se účastnilo 201 pacientů, byl porovnáván ambrisentan v dávce 10 mg a 5 mg s placebem. Ve studii ARIES-2, které se účastnilo 192 pacientů, byl porovnáván ambrisentan v dávce 2,5 mg a 5 mg s placebem. V obou studiích byl ambrisentan přidáván k podpůrné/původní léčbě, která mohla zahrnovat kombinaci digoxinu, antikoagulancií, diuretik, kyslíku a vazodilatancií (blokátory kalciového kanálu, ACE-inhibitory). Pacienti účastnící se studií měli IPAH nebo PAH spojenou s onemocněním pojivové tkáně (PAH-CTD). Většina pacientů měla příznaky II. stupně (38,4 %) nebo III. stupně (55,0 %) WHO funkční klasifikace. Pacienti s preexistujícím jaterním onemocněním (cirhóza nebo klinicky významně zvýšené hodnoty aminotransferáz) a pacienti užívající jinou cílenou léčbu PAH (např. prostanoidy) byli ze studie vyloučeni. Hemodynamické parametry nebyly v těchto studiích hodnoceny.
Primárním cílovým parametrem účinnosti definovaným pro fázi III klinických studií bylo zlepšení zátěžové kapacity, které bylo hodnoceno jako změna vzdálenosti 6 minutové chůze (6MWD) měřené ve 12. týdnu oproti stavu před léčbou. V obou studiích vedla léčba ambrisentanem ve všech podávaných dávkách k významnému zlepšení 6MWD.
Zlepšení průměrné 6MWD upravené k placebovému účinku při užívání dávky 5 mg činilo ve 12. týdnu ve srovnání s výchozí hodnotou 30,6 m (95 % IS: 2,9 až 58,3; p=0,008) ve studii ARIES-1 a 59,4 m (95% IS: 29,6 až 89,3; p<0,001) ve studii ARIES-2. Zlepšení průměrné 6MWD upravené k placebovému účinku při užívání dávky 10 mg činilo ve studii ARIES-1 51,4 m (95% IS: 26,6 až 76,2;
p <0,001).
Byla též provedena předem specifikovaná kombinovaná analýza výsledků studií fáze III (ARIES-C). Zlepšení průměrné 6MWD upravené k placebovému účinku činilo při užívání dávky 5 mg 44,6 m (95 % IS:24,3 až 64,9; p<0,001) a při užívání dávky 10 mg 52,5 m (95% IS: 28,8 až 76,2; p<0,001).
Ve studii ARIES-2 ambrisentan (ve skupině s kombinovanou dávkou) významně prodloužil dobu do klinického zhoršení PAH ve srovnání s placebem (p<0,001), došlo k 80% snížení rizika výskytu (95% IS: 47% až 92%) sledovaných faktorů. Hodnocení zahrnovalo: úmrtí, transplantaci plic, hospitalizaci pro PAH, atriální septostomii, přidání další medikace k léčbě PAH a kritéria pro předčasné ukončení léčby. Ve skupině s kombinovanou dávkou bylo ve srovnání s placebem pozorováno statisticky významné zlepšení (3,41 ± 6,96) fyzických funkcí hodnocených podle stupnice "SF-36 Health Survey" (-0,20 ± 8,14; p=0,005). Léčba ambrisentanem vedla ke statisticky významnému zlepšení indexu BDI ("Borg Dyspnoe Index") ve 12. týdnu (BDI upravený k placebovému účinku -1,1 (95% IS: -1,8 až -0,4; p=0,019; skupina s kombinovanou dávkou)).
Údaje z dlouhodobých klinických studií
Pacienti, kteří se účastnili studií ARIES 1 a 2, byli způsobilí účastnit se dlouhodobé otevřené rozšířené studie ARIES-E (n=383). Kombinovaná střední expozice léčivému přípravku byla přibližně 145 ± 80 týdnů a maximální expozice byla přibližně 295 týdnů. Hlavním primárním cílovým parametrem studie byla incidence a závažnost nežádoucích účinků souvisejících s dlouhodobou expozicí ambrisentanu, včetně sérových jaterních testů. Nálezy týkající se bezpečnosti pozorované při dlouhodobé expozici ambrisentanu v této studii byly obecně shodné s nálezy pozorovanými ve 12týdenních placebem kontrolovaných studiích.
Pozorovaná pravděpodobnost přežívání u subjektů léčených ambrisentanem (skupina s kombinovanou dávkou ambrisentanu) byla 93% po 1 roce léčby, 85% po 2 letech a 79% po 3 letech léčby.
V otevřené studii (AMB222) byl ambrisentan hodnocen u 36 pacientů a cílem studie bylo posouzení incidence zvýšených koncentrací aminotransferáz v séru u pacientů, u kterých došlo k předchozímu přerušení léčby jinými antagonisty receptorů pro endotelin z důvodu zvýšení koncentrací aminotransferáz. Během průměrné doby 53 týdnů léčby ambrisentanem nebylo u žádného z pacientů zaznamenáno zvýšení sérových koncentrací ALT >3x ULN, které by vyžadovalo trvalé ukončení léčby. U padesáti procent pacientů došlo během této doby ke zvýšení dávky ambrisentanu z 5 mg na 10 mg.
Kumulativní incidence zvýšených sérových koncentrací aminotransferáz >3x ULN ve všech studiích fáze 2 a 3 (včetně příslušné otevřené rozšířené studie) činila 17 subjektů ze 483 při průměrné délce expozice 79,5 týdnů. To odpovídá četnosti 2,3 příhody na 100 pacientoroků expozice ambrisentanu.
V otevřené dlouhodobé rozšířené studii ARIES-E bylo u pacientů léčených ambrisentanem dvouleté riziko rozvoje elevací sérových aminotransferáz >3x ULN 3,9%.
Další klinické údaje
Ve studii fáze 2 (AMB220) bylo po 12 týdnech léčby u pacientů s PAH (n=29) zaznamenáno zlepšení hemodynamických parametrů. Léčba ambrisentanem vedla ke zvýšení průměrného srdečního indexu, ke snížení průměrného tlaku v plicnici a ke snížení průměrné plicní cévní rezistence.
Při léčbě ambrisentanem byl hlášen pokles systolického a diastolického krevního tlaku. V placebem kontrolovaných klinických studiích trvajících 12 týdnů byl průměrný pokles systolického krevního tlaku od výchozích hodnot do ukončení léčby 3 mm Hg a diastolického krevního tlaku 4,2 mm Hg. Průměrný pokles systolického i diastolického krevního tlaku přetrvával po dobu až 4 let během léčby ambrisentanem v dlouhodobé otevřené studii ARIES-E.
V lékové interakční studii u zdravých dobrovolníků nebyl zaznamenán klinicky významný vliv na farmakokinetiku ambrisentanu nebo sildenafilu a tato kombinace byla dobře snášena. Počet pacientů, kteří užívali ambrisentan současně se sildenafilem, činil ve studii ARIES-E 22 pacientů (5,7 %) a ve studii AMB222 17 pacientů (47 %). Žádné další problémy vztahující se k bezpečnosti nebyly u těchto pacientů zaznamenány.
Klinická účinnost v kombinaci s tadalafilem
Multicentrická, dvojitě zaslepená, událostí řízená studie 3. fáze s aktivním komparátorem (AMB112565/AMBITION) byla provedena pro posouzení účinnosti počátečního kombinace ambrisentanu a tadalafilu versus monoterapie buď ambrisentanem nebo tadalafilem samotným na 500 dosud neléčených pacientech s PAH, randomizovaných v poměru 2:1:1. Žádný z pacientů nedostával pouze placebo. Primární analýza hodnotila skupinu s kombinací oproti poolovaným datům skupin s monoterapií. Podpůrné srovnání skupiny s kombinovanou léčbou versus jednotlivé skupiny s monoterapií bylo též provedeno. Pacienti s výraznou anémií, retencí tekutin nebo vzácnými chorobami sítnice byli vyloučeni v souladu s kritérii investigátorů. Pacienti s hodnotami ALT a AST > dvojnásobku horní hranice normy byli rovněž vyloučeni.
Při vstupu do studie, 96% pacientů bylo naivních s ohledem na jakoukoli předchozí PAH specifickou léčbu a střední doba od diagnózy do vstupu do studie byla 22 dní. Pacienti, kteří začínali s ambrisentanem 5 mg a tadalafilem 20 mg byli titrováni na 40 mg tadalafilu ve 4. týdnu a 10 mg ambrisentanu v 8. týdnu, pokud nebyly problémy se snášenlivostí. Střední doba dvojitě zaslepené léčby pro kombinovanou terapii byla vyšší než 1,5 roku.
Primárním cílovým parametrem byl čas do prvního výskytu klinického selhání, definovaného jako:
- úmrtí, nebo
- hospitalizace kvůli zhoršení PAH,
- progrese onemocnění;
- dlouhodobá nevyhovující klinická odpověď.
Střední věk všech pacientů byl 54 let (SD 15, rozpětí 18-75 let). Pacienti byli na začátku studie ve II. (31%) a III. (69%) funkční třídě dle klasifikace WHO. Idiopatická nebo dědičná PAH byla nejčastější etiologií ve studované populaci (56%), následovaná PAH v důsledku poškození pojivové tkáně (37%), PAH související s léčivy a toxiny (3%), korigované nekomplikované vrozené srdeční vady (2% ) a HIV (2%). Pacienti s PAH II. a III. třídy dle klasifikace WHO měli střední výchozí 6MWD 353 metrů.
Výsledky hodnocení
Léčba kombinovanou terapií měla za následek 50% snížení rizika (poměr rizika [HR] 0,502; 95% CI: 0,384 až 0,724, p = 0,0002) složeného parametru účinnosti klinického selhání až do poslední návštěvy ve srovnání s monoterapií [Obrázek 1 a Tabulka 1]. Efekt léčby spočívající v 63% snížení nutnosti hospitalizace při kombinované terapii byl zaznamenán brzy a byl zachován během léčby. Účinnost kombinované terapie vzhledem k primárnímu cíli byla konzistentní ve srovnání s individuální monoterapií a napříč podskupinami podle věku, etnického původu, zeměpisné oblasti, etiologie (IPAH / hPAH a PAH-CTD). Účinek byl významný pro pacienty jak II. tak i III. funkční třídy dle klasifikace WHO.
Obrázek 1
Čas do klinického selhání
Počet v riziku
|
fct-=>=_. 1 rok 88,9% | |
|
| |
2 roky 79,7 % í |
|
^-i ? rnkv P7 fí % | |
|
* | |
|
1 rok 75,5 % |
_ |
|
• “ “ ” “ i _ : | |
|
2 roky 63,2% -------- | |
|
: 3 roky 56,1% | |
|
— Kombinovaná terapie |
i PR: 0,502 |
|
: 95% Cl (0,348; 0,724) | |
|
- - Monoterapie poolovaná |
i p=0,0002 |
24 48 72 96 120 144 168 192
Čas (týdny)
Kombinace:
253 229 186 145 106 71 36
Monoterapie poolovaná: 247 209 155 108 77 49 25
Tabulka 1
|
Ambrisentan + Tadalafil (N=253) |
Monoterapie Poolovaná data (N=247) |
Ambrisentan monoterapie (N=126) |
Tadalafil monoterapie (N=121) | |
|
Čas do události -prvního klinického selhání (určeno) | ||||
|
Klinické selhání, počet (%) |
46 (18%) |
77 (31%) |
43 (34) |
34 (28) |
|
Poměr rizika (95% CI) |
0,502 (0,348 -0,724) |
0,477 (0,314 - 0,723) |
0,528 (0,338 - 0,827) | |
|
Hodnota p, Log-rank test |
0,0002 |
0,0004 |
0,0045 | |
|
Jednotlivé události stanovené jako událost prvního klinického selhání (určeno) | ||||
|
Úmrtí (všechny příčiny) |
9 (4%) |
8 (3%) |
2 (2) |
6 (5) |
|
Hospitalizace pro zhoršení PAH |
10 (4%) |
30 (12%) |
18 (14) |
12 (10) |
|
Progrese nemoci |
10 (4%) |
16 (6%) |
12 (10) |
4 (3) |
|
Dlouhodobě neuspokojivá klinická odpověď |
17 (7%) |
23 (9%) |
11 (9) |
12 (10) |
|
Čas do první hospitalizace pro zhoršení PAH (určeno) | ||||
|
První hospitalizace, časový údaj (%) |
19 (8%) |
44 (18%) |
27 (21%) |
17 (14%) |
|
Poměr rizika (95% CI) |
0.372 |
0.323 |
0.442 | |
|
Hodnota p, Log-rank test |
0.0002 |
<0.0001 |
0.0124 | |
Sekundární_parametry účinnosti
Jako sekundární parametry účinnosti byly testovány:
Tabulka 2
|
Sekundární parametry účinnosti (změna od zahájení studie do 24 týdne) |
Ambrisentan + Tadalafil |
Monoterapie- Poolovaná data |
Rozdíl a interval spolehlivosti |
hodnota p |
|
NT-proBNP (% snížení) |
-67,2 |
-50,4 |
% rozdíl -33,8; 95% CI: -44,8, -20,7 |
p<0.0001 |
|
% subjektů dosahujících uspokojivé klinické odpovědi ve 24. týdnu |
39 |
29 |
Poměr pravděpodobnosti 1,56; 95% CI: 1,05, |
p=0,026 |
|
2,32 | ||||
|
6MWD (metry, střední změna) |
49,0 |
23,8 |
22.75m; 95% CI: 12,00; 33,50 |
p<0,0001 |
Idiopatická plicní fibróza
Byla provedena studie hodnotící 492 pacientů (ambrisentan n = 329, placebo n = 163) s idiopatickou plicní fibrózou (IPF), z nichž 11 % mělo sekundární plicní hypertenzi (WHO skupina 3). Studie ale byla ukončena předčasně, když bylo zjištěno, že primární cílový parametr účinnosti nemůže být splněn (studie ARTEMIS-IPF). Ve skupině s ambrisentanem bylo pozorováno 90 případů (27 %) progrese IPF (včetně hospitalizace z důvodu zhoršení respirace) nebo úmrtí ve srovnání s 28 případy (17 %) ve skupině s placebem. Ambrisentan je proto kontraindikován u pacientů s IPF se sekundární plicní hypertenzí nebo bez ní (viz bod 4.3).
5.2 Farmakokinetické vlastnosti
Absorpce
Ambrisentan se u lidí rychle vstřebává. Po perorálním podání je maximálních plazmatických koncentrací ambrisentanu (Cmax) dosaženo přibližně za 1,5 hodiny po užití dávky nalačno i po jídle. Cmax a plocha pod křivkou plazmatických koncentrací (AUC) se v léčebném dávkovém rozmezí zvyšují úměrně s dávkou. Ustáleného stavu je obvykle dosaženo za 4 dny při opakovaném podávání.
U zdravých dobrovolníků, kteří užívali ambrisentan nalačno nebo s jídlem s vysokým obsahem tuků, byla provedena studie hodnotící možný vliv potravy. V této studii došlo ke snížení Cmax o 12 % zatímco AUC zůstala nezměněna. Toto snížení maximální plazmatické koncentrace není klinicky významné a ambrisentan proto lze užívat jak nalačno, tak s jídlem.
Distribuce v organismu
Ambrisentan se ve vysoké míře váže na plazmatické proteiny. In vitro činila vazba ambrisentanu na plazmatické proteiny v průměru 98,8 % a v rozmezí 0,2 - 20 mikrogramů/ml byla nezávislá na koncentraci. Ambrisentan je vázán zejména na albumin (96,5 %) a v menší míře na alfa1-kyselý glykoprotein.
Distribuce ambrisentanu do erytrocytů je nízká, průměrný poměr krev:plazma činí 0,57 u mužů a 0,61 u žen.
Biotransformace
Ambrisentan je nesulfonamidový ERA (kyselina propionová).
Ambrisentan podléhá glukuronidaci pomocí izoenzymů UGT (UGT1A9S, UGT2B7S a UGT1A3S) za vzniku ambrisentan-glukuronidu (13 %). Ambrisentan je také přeměňován cestou oxidativního metabolismu zejména působením CYP3A4 a v menší míře CYP3A5 a CYP2C19 za vzniku 4-hydroxymethyl-ambrisentanu (21 %), který je dále glukuronidován na 4-hydroxymethyl-ambrisentan glukuronid (5 %). Vazebná afinita 4-hydroxymethyl-ambrisentanu k lidskému receptoru pro endotelin je 65x nižší než u ambrisentanu. Nepředpokládá se tedy, že by 4-hydroxymethyl-ambrisentan v koncentracích zaznamenaných v plazmě (přibližně 4 % koncentrace mateřské látky) přispíval k farmakologické aktivitě ambrisentanu.
M
Údaje in vitro ukazují, že ambrisentan při koncentraci 300 pM vedl k méně než 50% inhibici UGT1A1, UGT1A6, UGT1A9, UGT2B7 (až do 30 %) nebo izoenzymů 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 a 3A4 cytochromu P450 (až 25 %). In vitro, ambrisentan v klinicky relevantních koncentracích nemá inhibiční účinek na lidské transportéry, včetně Pgp, BCRP, MRP2, BSEP, OATP1B1, OATP1B3 a NTCP. Navíc, ambrisentan neindukuje expresi proteinů MRP2, Pgp nebo BSEP v potkaních hepatocytech. Celkově vzato, in vitro data naznačují, že nelze předpokládat, že by ambrisentan v klinicky relevantních koncentracích (Cmax do 3,2 pM v plazmě) ovlivňoval UGT1A1, UGT1A6, UGT1A9, UGT2B7 nebo izoenzymy 1A2, 2A6, 2B6, 2C8, 2C9 , 2C19, 2D6, 2E1, 3A4 cytochromu P450 nebo transport přes BSEP, BCRP, PGP, MRP2, OATP1B1 / 3, nebo NTCP.
Vliv ambrisentanu v ustáleném stavu (10 mg jedenkrát denně) na farmakokinetiku a farmakodynamiku jednorázově podaného warfarinu (25 mg) byl hodnocen pomocí PT a INR u 20 zdravých dobrovolníků. Ambrisentan neměl klinicky významný účinek na farmakokinetiku nebo farmakodynamiku warfarinu. Podobně současné podávání warfarinu neovlivňovalo farmakokinetiku ambrisentanu (viz bod 4.5).
U 19 zdravých dobrovolníků byl hodnocen vliv podávání sildenafilu po dobu 7 dnů (20 mg třikrát denně) na farmakokinetiku ambrisentanu při jednorázovém podání a vliv podávání ambrisentanu po dobu 7 dnů (10 mg jedenkrát denně) na farmakokinetiku sildenafilu při jednorázovém podání. Kromě 13% zvýšení Cmax sildenafilu nedošlo při současném podání s ambrisentanem k žádným jiným změnám farmakokinetických parametrů sildenafilu, N-desmethyl-sildenafilu ani ambrisentanu. Toto mírné zvýšení Cmax sildenafilu není považováno za klinicky významné (viz bod 4.5).
U 23 zdravých dobrovolníků byl hodnocen vliv ambrisentanu v ustáleném stavu (10 mg jedenkrát denně) na farmakokinetiku tadalafilu po jednorázovém podání a vliv tadalafilu v ustáleném stavu (40 mg jedenkrát denně) na farmakokinetiku ambrisentanu po jednorázovém podání. Ambrisentan neměl klinicky významný účinek na farmakokinetiku tadalafilu. Podobně současné podávání s tadalafilem neovlivnilo farmakokinetiku ambrisentanu (viz bod 4.5).
U 16 zdravých dobrovolníků byl hodnocen vliv ketokonazolu při opakovaném podávání (400 mg jedenkrát denně) na farmakokinetiku ambrisentanu při jednorázovém podání 10 mg. Expozice ambrisentanu měřené pomocí AUC^jf a Cmax byly zvýšeny o 35 %, respektive o 20 %. Je nepravděpodobné, že by tyto změny v expozici měly klinický význam, a proto může být ambrisentan podáván současně s ketokonazolem.
Účinky opakovaných dávek cyklosporinu A (100 - 150 mg dvakrát denně) na farmakokinetiku ambrisentanu v rovnovážném stavu (5 mg jednou denně) a účinky opakovaných dávek ambrisentanu (5 mg jednou denně) na farmakokinetiku cyklosporinu A v rovnovážném stavu (100 - 150 mg dvakrát denně) byly studovány na zdravých dobrovolnících. Cmax a AUC(0-t) ambrisentanu při mnohočetných dávkách cyklosporinu A stoupalo (48 % resp. 121 %). Na základě těchto změn by dávka ambrisentanu měla být omezena na 5 mg jednou denně, pokud je podáván společně s cyklosporinem A (viz bod 4.2). Mnohočetné dávky ambrisentanu nemají na expozici cyklosporinu A žádný vliv, a proto žádná úprava dávky cyklosporinu A není nutná.
U zdravých dobrovolníků byly studovány účinky akutních a opakovaných dávek rifampicinu (600 mg jedenkrát denně) na farmakokinetiku ambrisentanu (10 mg jedenkrát denně) v ustáleném stavu. Po počátečních dávkách rifampicinu bylo pozorováno přechodné zvýšení hodnoty AUC(0-T) ambrisentanu (121% po první dávce rifampicinu a 116% po druhé dávce rifampicinu), pravděpodobně v důsledku inhibice OATP zprostředkované rifampicinem. Avšak, po podání vícenásobných dávek rifampicinu nebyl do osmého dne pozorován žádný klinicky významný vliv na expozici ambrisentanu. Pacienti, kterým je podáván ambrisentan, musí být po zahájení léčby rifampicinem pečlivě sledováni (viz bod 4.5 a 4.5).
U 15 zdravých dobrovolníků byl hodnocen vliv ambrisentanu (10 mg) při opakovaném podávání na farmakokinetiku digoxinu při jednorázovém podání. Opakované podání ambrisentanu vedlo k mírnému zvýšení AUC0-last a nejnižších koncentrací digoxinu a k 29% zvýšení Cmax digoxinu. Zvýšení expozice digoxinu pozorované při současném podávání opakovaných dávek ambrisentanu nebylo považováno za klinicky významné a úprava dávkování digoxinu tedy není nutná (viz bod 4.5).
U zdravých dobrovolnic byl hodnocen vliv 12denního podávání ambrisentanu (10 mg jedenkrát denně) na farmakokinetiku perorálního antikoncepčního přípravku obsahujícího ethinylestradiol (35gg) a norethisteron (1 mg) po jednorázovém podání. Cmax a AUC(0-X)) ethinylestradiolu byly mírně sníženy (o 8 %, resp. o 4 %) a Cmax a AUC(0-<X)) norethisteronu byly mírně zvýšeny (o 13 %, resp. o 14 %). Tyto změny expozice ethinylestradiolu nebo norethisteronu byly malé a je nepravděpodobné, že by mohly být klinicky významné (viz bod 4.5).
Eliminace z organismu
Ambrisentan a jeho metabolity jsou po hepatální a/nebo extrahepatální metabolizaci vylučovány zejména žlučí. Po perorálním podání se přibližně 22 % podané dávky vyloučí močí; 3,3 % ve formě nezměněného ambrisentanu. Plazmatický eliminační poločas je u lidí v rozmezí 13,6 až 16,5 hodiny.
Zvláštní skupiny pacientů
Výsledky populační farmakokinetické analýzy u zdravých dobrovolníků a pacientů s PAH prokázaly, že farmakokinetika ambrisentanu není významně ovlivněna pohlavím ani věkem (viz bod 4.2).
Porucha funkce ledvin
Ambrisentan nepodléhá významnému renálnímu metabolismu ani renální clearance (vylučování). V populační farmakokinetické analýze byla clearance kreatininu statisticky významnou veličinou ovlivňující clearance ambrisentanu po perorálním podání. Snížení clearance po perorálním podání je u pacientů se středně závažným renálním poškozením mírné (20-40 %) a je tedy nepravděpodobné, že by mohlo být klinicky významné. U pacientů se závažným poškozením ledvin je však nutná opatrnost (viz bod 4.2).
Porucha funkce jater
Hlavní metabolickou cestou ambrisentanu je glukuronidace a oxidace s následným vylučováním žlučí a mohlo by se tedy předpokládat, že jaterní poškození může zvýšit expozici (Cmax a AUC) ambrisentanu. V populační farmakokinetické analýze se clearance po perorálním podání snižovala v závislosti na zvyšujících se hladinách bilirubinu. Vliv bilirubinu je však mírný (ve srovnání s pacientem s běžnou hodnotou bilirubinu 0,6 mg/dl by měl pacient s hladinou bilirubinu zvýšenou na
4,5 mg/dl přibližně o 30 % nižší clearance ambrisentanu po perorálním podání). Farmakokinetika ambrisentanu nebyla u pacientů s jaterním poškozením (s cirhózou nebo bez cirhózy) hodnocena. Léčba ambrisentanem se proto nemá zahajovat u pacientů s poruchou funkce jater nebo s klinicky významně zvýšenými hodnotami jaterních aminotransferáz (>3x ULN) (viz body 4.3 and 4.4).
5.3 Předklinické údaje vztahující se k bezpečnosti
Vzhledem k hlavnímu farmakologickému účinku této třídy léčiv by mohlo vést podání vysoké jednorázové dávky ambrisentanu (tj. předávkování) ke snížení krevního tlaku a k možné hypotenzi a příznakům souvisejícím s vazodilatací.
Nebylo prokázáno, že by ambrisentan inhiboval transport žlučových kyselin nebo že by byl významně hepatotoxický.
Po dlouhodobém podávání byly u hlodavců v expozicích nižších, než odpovídají terapeutickým hladinám u člověka, pozorovány zánět a změny epitelu nosní dutiny. U psů byla při dlouhodobém podávání vysokých dávek ambrisentanu v expozicích vyšších než 20násobek expozice u člověka pozorována mírná zánětlivá odpověď.
U potkanů, kterým byl podáván ambrisentan v expozicích 3krát vyšších, než jsou klinické hodnoty AUC, byla v nosní dutině zaznamenána hyperplazie nosní kosti (kosti čichové). Hyperplazie nosní kosti nebyla při podávání ambrisentanu pozorována u myší a psů. Na základě zkušeností s dalšími sloučeninami je hyperplazie nosní skořepy u potkanů zjevnou odpovědí na nosní zánět.
Při testování in vitro na buňkách savců byl ambrisentan ve vysokých koncentracích klastogenní. Mutagenní ani genotoxický účinek ambrisentanu nebyl v testech na bakteriích ani ve dvou in vivo studiích na hlodavcích prokázán.
Ve 2letých studiích perorálního podávání u potkanů a myší nebyl prokázán karcinogenní potenciál. Byl zaznamenán malý nárůst incidence fibroadenomů prsu (benigních tumorů) a to pouze u samců potkanů při nejvyšší dávce. Systémová expozice ambrisentanu byla u samců potkanů při této dávce (na základě AUC v ustáleném stavu) 6násobná ve srovnání s expozicí, které je dosahováno při klinické dávce 10 mg/den.
Ve studiích toxicity při opakovaném perorálním podávání a ve studiích fertility provedených na potkaních a myších samcích mimo bezpečnostní rozpětí byla zaznamenána testikulární tubulární atrofie, která byla v některých případech spojena s aspermií. V průběhu hodnoceného období bez podávání dávek se testikulární změny zcela neupravily. Nicméně ve studiích na psech, trvajících až 39 týdnů při expozici 35krát vyšší než je expozice u lidí určená na základě AUC, nebyly pozorovány žádné testikulární změny. Při žádné z testovaných dávek (až 300 mg/kg/den) nebyly u samců potkanů pozorovány žádné účinky ambrisentanu na motilitu spermií. Při dávkách 300 mg/kg/den bylo pozorováno mírné (<10 %) snížení procenta morfologicky normálních spermií; při dávkách 100 mg/kg/den (> 9násobek klinické expozice 10 mg/den) však toto snížení pozorováno nebylo.
Vliv ambrisentanu na fertilitu u mužů není znám.
Ambrisentan měl u potkanů a králíků teratogenní účinky. Anomálie dolní čelisti, jazyka a/nebo patra byly pozorovány ve všech testovaných dávkách. Ve studiích na potkanech byl navíc zaznamenán zvýšený výskyt defektů komorového septa, defektů kmenových cév, anomálií štítné žlázy a thymu, osifikace basisphenoidální kosti a lokalizace umbilikální arterie na levé straně močového měchýře místo na pravé straně.
Teratogenita je očekávaným nežádoucím účinkem celé třídy antagonistů receptorů pro endotelin.
Při expozicích 3krát převyšujících AUC maximálních doporučených dávek u lidí, vedlo podávání ambrisentanu potkaním samicím v období od pozdní březosti po laktaci k nežádoucímu ovlivnění mateřského chování, ke sníženému přežívání mláďat a k poškození reprodukční schopnosti potomstva (v pitevním nálezu byla pozorována malá varlata).
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety Monohydrát laktosy Mikrokrystalická celulosa Sodná sůl kroskarmelosy Magnesium-stearát
Potah tablety
Částečně hydrolyzovaný polyvinylalkohol Mastek (E553b)
Oxid titaničitý (E171)
Makrogol/PEG 3350
Sójový lecithin (E322)
Hlinitý lak červeně Allura AC (E129)
6.2 Inkompatibility
Neuplatňuje se
6.3 Doba použitelnosti
2 roky.
6.4 Zvláštní opatření pro uchovávání
Tento léčivý přípravek nevyžaduje žádné zvláštní podmínky uchovávání.
6.5 Druh obalu a obsah balení
PVC/PVDC/Al blistry. Velikost balení s jednodávkovými blistry po 10x1 nebo 30x1 potahovaných tabletách. Na trhu nemusí být k dispozici všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky pro likvidaci.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd
980 Great West Road
Brentford
Middlesex
TW8 9GS
Velká Británie
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/08/451/003
EU/1/08/451/004
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 21. dubna 2008 Datum posledního prodloužení: 24.dubna 2013
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách
Evropské agentury pro léč ivé př ípravky na adrese http://www.ema.europa.eu.
PŘÍLOHA II
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
Název a adresa výrobce odpovědného za propouštění šarží
Aspen Bad Oldesloe GmbH Industriestrasse 32-36 D-23843 Bad Oldesloe Německo
nebo
Glaxo Operations UK Ltd
(trading as GlaxoWellcome Operations)
Harmire Road Barnard Castle Co. Durham DL12 8DT Velká Británie
V tištěné příbalové informaci k léčivému přípravku musí být uveden název a adresa výrobce zodpovědného za propuštění konkrétní šarže přípravku.
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz Příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ TOHOTO LÉČIVÉHO PŘÍPRAVKU
Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Další opatření k minimalizaci rizik
Držitel rozhodnutí o registraci si musí nechat odsouhlasit národním regulačním úřadem podrobnosti systému řízené distribuce a zavede tento systém na národní úrovni tak, aby byly všem zdravotnickým pracovníkům, kteří budou předepisovat přípravek Volibris (a kde je to vhodné a po schválení národním regulačním úřadem event. vydávat) ještě před předepsáním (případně výdejem) poskytnuty následující materiály:
• Informace o přípravku (Souhrn údajů o přípravku (SPC) a příbalová informace)
• Informace o přípravku Volibris určená pro zdravotnické pracovníky
• Kontrolní seznam určený pro lékaře před předepsáním léčby
• Informace o studii postmarketinkového sledování
• Informační brožurky pro pacienty
• Informační brožurky pro partnery pacientek v reprodukčním věku
• Připomínkové karty pacienta (“Patient reminder cards”)
• Formuláře na hlášení těhotenství
• Formuláře na hlášení nežádoucích účinků
Informace pro zdravotnické pracovníky
Informace o přípravku Volibris určená pro zdravotnické pracovníky musí obsahovat následující klíčové informace:
Povinnosti zdravotnických pracovníků při předepisování přípravku Volibris:
• Zajistit, že pacienti jsou schopni dodržovat požadavky na bezpečné užití přípravku Volibris.
• Poskytování srozumitelných rad a informací pacientům.
• Předání příslušných informačních brožurek a připomínkové karty pacientům.
• Zvážit měsíční předepisování na 30denní léčbu, aby bylo zajištěno, že pacienti byli vyšetřeni a výsledky důležitých testů byly zhodnoceny před další preskripcí léčiva.
• Vzhledem k tomu, že je databáze údajů o bezpečnosti přípravku Volibris omezená, jsou lékaři vybízeni k zařazování pacientů do studie postmarketinkového sledování.
• Hlásit suspektní nežádoucí účinky a těhotenství.
Přípravek Volibris je teratogenní
• Podávání přípravku Volibris je kontraindikováno v těhotenství a u žen v reprodukčním věku, které nepoužívají účinnou antikoncepci.
• Ženy užívající přípravek Volibris musí být informovány o riziku poškození plodu.
• Pokyny k identifikaci žen v reprodukčním věku a informace, jaké kroky učinit, pokud si lékař není jistý.
Pokyny týkající se žen v reprodukčním věku:
• Vyloučení těhotenství před zahájením léčby a provádění těhotenských testů jedenkrát měsíčně.
• Nutnost doporučit ženám (a to i ženám s amenoreou) používat v průběhu léčby a ještě jeden měsíc po ukončení léčby účinnou antikoncepci.
• Definice účinné antikoncepce a nutnost porady s příslušným specialistou v případě pochybností o vhodné antikoncepci pro konkrétní pacientku.
• Pokud žena v reprodukčním věku potřebuje změnit nebo ukončit používání antikoncepce musí informovat lékaře předepisujícího antikoncepci o tom, že užívá přípravek Volibris .
• Pokud žena v reprodukčním věku potřebuje změnit nebo ukončit používání antikoncepce musí informovat lékaře předepisujícího přípravek Volibris.
• Pacientka musí neprodleně informovat svého lékaře při podezření na možné těhotenství a v případě potvrzení těhotenství musí být zahájena alternativní léčba.
• Nutnost odeslat pacientku, která otěhotněla, k lékaři se specializací nebo zkušenostmi v oblasti teratologie k posouzení diagnózy a získání doporučení k dalšímu postupu.
• Hlásit všechny případy těhotenství, ke kterým dojde během léčby.
Volibris je potenciálně hepatotoxický
• Kontraindikace u pacientů s těžkou poruchou funkce jater (s cirhózou nebo bez cirhózy) a u pacientů s výchozími hodnotami jaterních aminotransferáz (AST a/nebo ALT) > 3x ULN.
• Hodnoty jaterních aminotransferáz (ALT a/nebo AST) musí být posouzeny před zahájením léčby ambrisentanem.
• Během léčby se doporučuje měsíčně kontrolovat hodnoty ALT a AST.
• Přerušení léčby ambrisentanem, pokud dojde u pacienta k rozvoji přetrvávajícího, nejasného, klinicky významného zvýšení hodnot ALT a/nebo AST nebo je-li zvýšení hodnot ALT a/nebo AST doprovázeno známkami nebo příznaky jaterního poškození (např. žloutenka).
• U pacientů bez klinických příznaků jaterního poškození nebo žloutenky může být po úpravě hodnot jaterních enzymů zváženo opětovné zahájení podávání ambrisentanu. Doporučuje se konzultace s hepatologem.
Léčba přípravkem Volibris často způsobuje snížení hemoglobinu a hematokritu
• U pacientů s klinicky významnou anémií se zahájení léčby přípravkem Volibris nedoporučuje.
• U pacientů užívajících přípravek Volibris mají být hodnoty hemoglobinu a/nebo hematokritu monitorovány v pravidelných intervalech.
• Pokud testy prokáží klinicky významné snížení hodnot hemoglobinu nebo hematokritu a jiné příčiny jsou vyloučeny, mělo by být zváženo snížení dávky nebo přerušení léčby přípravkem Volibris.
Léčba přípravkem Volibris může způsobit periferní otok a retenci tekutin
• Dojde-li k rozvoji klinicky významného periferního otoku, s nárůstem tělesné hmotnosti nebo bez něj, je nutno dále posoudit příčinu tohoto stavu a v případě nutnosti zvážit přerušení léčby přípravkem Volibris.
Dlouhodobé podávání přípravku Volibris u zvířat bylo spojeno s rozvojem tubulární atrofie varlat a poruchou fertility. Vliv přípravku Volibris na testikulární funkci u lidí a na fertilitu u mužů není znám.
U pacientů s těžkou poruchou funkce ledvin má být léčba přípravkem Volibris zahájena s opatrností.
Při léčbě přípravkem Volibris byly hlášeny reakce přecitlivělosti (např. angioedém, vyrážka) a to méně často v krátkodobých klinických studiích a často v dlouhodobých studiích s tadalafilem.
Kontrolní seznam pro lékaře
Kontrolní seznam před zahájením léčby určený pro lékaře upozorňuje na kontraindikace podání ambrisentanu a obsahuje důležitá posouzení, která je třeba učinit před zahájením léčby zahrnující:
• Posouzení funkce jater (j aterní testy).
• Stanovení, zda je pacientka v reprodukčním věku.
• Provedení těhotenského testu u pacientek v reprodukčním věku.
• Posouzení, zda pacientky v reprodukčním věku používají účinnou antikoncepci.
Informace pro pacienta
Informace pro pacienta musí obsahovat následující údaje:
• Přípravek Volibris může způsobit závažné vrozené vady nenarozeného dítěte, které bylo počato před léčbou, v průběhu léčby nebo v průběhu jednoho měsíce po ukončení léčby.
• Pokud j e pacientka těhotná, léčba přípravkem Volibris nemůže být zaháj ena.
• U žen v reprodukčním věku musí být proveden těhotenský test bezprostředně před zahájením léčby a dále v průběhu léčby přípravkem Volibris v měsíčních intervalech.
• Nutnost zajistit, aby žena v reprodukčním věku používala účinnou antikoncepci a aby informovala své lékaře v případě podezření na možnost těhotenství předtím, než je vydán nový recept na další léčbu.
• Pokud žena v reprodukčním věku potřebuje změnit nebo ukončit používání antikoncepce musí informovat lékaře předepisujícího antikoncepci o tom, že užívá přípravek Volibris .
• Pokud žena v reprodukčním věku potřebuje změnit nebo ukončit používání antikoncepce musí informovat lékaře předepisujícího přípravek Volibris.
• V případě podezření na možnost těhotenství musí pacientka neprodleně informovat svého ošetřujícího lékaře.
• Pokud pacientka plánuje těhotenství, musí o tom informovat svého lékaře.
• Volibris může způsobit poškození jater.
• Vzhledem k nebezpečí možného poškození jater a rozvoje anémie musí pacienti v pravidelných intervalech podstupovat krevní testy a musí oznámit svému lékaři jakékoli zaznamenané příznaky jaterního poškození.
• Přípravek Volibris nesmějí užívat žádné jiné osoby.
• Je třeba, aby pacient oznámil svému lékaři, pokud zaznamená jakékoli nežádoucí účinky.
Informační brožurka pro partnery pacientek v reprodukčním věku
Informační brožurka pro partnery pacientek v reprodukčním věku musí obsahovat následující
informace:
• Přípravek Volibris může způsobit závažné vrozené vady nenarozeného dítěte, které bylo počato před léčbou, v průběhu léčby nebo v průběhu jednoho měsíce po ukončení léčby.
• Nutnost zajistit, aby žena v reprodukčním věku používala účinnou antikoncepci.
• Přípravek Volibris nesmějí užívat těhotné ženy nebo ženy, které by mohly otěhotnět.
Připomínková karta pacienta
Musí obsahovat důležitá sdělení týkající se nutnosti pravidelného provádění krevních a těhotenských
testů a musí obsahovat dostatečné místo pro zaznamenávání dat, na kdy je pacient objednán k lékaři a
výsledků laboratorních vyšetření.
• <Povinnost uskutečnit poregistrační opatření>
Neuplatňuje se.
PŘÍLOHA III
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Volibris 5 mg potahované tablety ambrisentanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ambrisentanum 5 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu, sójový lecithin (E322) a hlinitý lak červeně Allura AC (E129). Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10x1 potahovaných tablet.
30x1 potahovaných tablet.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.
7. DALŠÍ ZVLÁŠTNÍ UPOZORNĚNÍ, POKUD JE POTŘEBNÉ
8. POUŽITELNOST
EXP
9. ZVLÁŠTNÍ PODMÍNKY PRO UCHOVÁVÁNÍ
11. NÁZEV A ADRESA DRŽITELE ROZHODNUTÍ O REGISTRACI
Glaxo Group Ltd 980 Great West Road Brentford
Middlesex TW8 9GS Velká Británie
12. REGISTRAČNÍ ČÍSLO/ČÍSLA
EU/1/08/451/001 10 potahovaných tablet EU/1/08/451/002 30 potahovaných tablet
13. ČÍSLO ŠARŽE
č.š.:
14. KLASIFIKACE PRO VÝDEJ
15. NÁVOD K POUŽITÍ
16. INFORMACE V BRAILLOVĚ PÍSMU
Volibris 5 mg
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Volibris 10 mg potahované tablety ambrisentanum
2. OBSAH LÉČIVÉ LÁTKY/LÉČIVÝCH LÁTEK
Jedna tableta obsahuje ambrisentanum 10 mg.
3. SEZNAM POMOCNÝCH LÁTEK
Obsahuje laktosu, sójový lecithin (E322) a hlinitý lak červeně Allura AC (E129). Další informace viz příbalová informace.
4. LÉKOVÁ FORMA A OBSAH BALENÍ
10x1 potahovaných tablet.
30x1 potahovaných tablet.
5. ZPŮSOB A CESTA/CESTY PODÁNÍ
Před použitím si přečtěte příbalovou informaci. Perorální podání.
6. ZVLÁŠTNÍ UPOZORNĚNÍ, ŽE LÉČIVÝ PŘÍPRAVEK MUSÍ BÝT UCHOVÁVÁN MIMO DOHLED A DOSAH DĚTÍ
Uchovávejte mimo dohled a dosah dětí.