Pixuvri 29 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Pixuvri 29 mg prášek pro koncentrát pro infuzní roztok
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna injekční lahvička obsahuje pixantron-dimaleinát, v množství, které odpovídá 29 mg pixantronum.
Po rekonstituci jeden ml koncentrátu obsahuje pixantron-dimaleinát v množství, které odpovídá 5,8 mg pixantronum.
Pomocná látka se známým účinkem:
Jedna injekční lahvička obsahuje 39 mg sodíku.
Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Prášek pro koncentrát pro infuzní roztok.
Tmavě modrý lyofilizovaný prášek.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Pixuvri je indikován jako monoterapie k léčbě dospělých pacientů s opakovaně relabujícími nebo refrakterními agresivními non-hodgkinskými lymfomy z B-buněk (NHL). Přínos léčby pixantronem jakožto léčby páté nebo vyšší linie chemoterapie u pacientů, kteří nereagovali na poslední léčbu, nebyl dosud stanoven.
4.2 Dávkování a způsob podání
Přípravek Pixuvri musí podávat lékaři, kteří jsou dobře obeznámeni s použitím antineoplastických látek a mají vybavení k pravidelnému sledování klinických, hematologických a biochemických parametrů v průběhu léčby a po ní (viz bod 6.6).
Dávkování
Doporučená dávka je 50 mg/m2 pixantronu v 1., 8. a 15. dni každého 28denního cyklu po dobu 6 cyklů.
Všimněte si:
Doporučená dávka v EU se vztahuje k bázi léčivé látky (pixantronu). Výpočet konkrétní dávky, kterou je třeba pacientovi podat, musí vycházet ze síly rekonstituovaného roztoku, který obsahuje 5,8 mg/ml pixantronu, a z doporučené dávky 50 mg/m2 . V některých studiích a publikacích doporučená dávka vychází z formy soli (pixantron-dimaleinát).
Dávku však je třeba před zahájením každého cyklu upravit na základě hodnot krevního obrazu v době
jejich nejhlubšího poklesu (nadir) nebo na základě maximální toxicity zjištěné v předchozím cyklu terapie. Množství přípravku Pixuvri v miligramech, které se má pacientovi podat, by se mělo určit na základě plochy povrchu těla pacienta. Plocha povrchu těla by se měla stanovit pomocí institucionální normy pro jeho výpočet a měla by se při tom použít tělesná hmotnost naměřená v 1. den každého cyklu.
U obézních pacientů se doporučuje postupovat s jistou obezřetností, neboť údaje o dávkování na
2
základě plochy tělesného povrchu jsou u této skupiny velmi omezené.
Pokyny k úpravě dávkování
Úprava dávkování a načasování následných dávek by se měly určit klinickým posouzením v závislosti na stupni a délce trvání myelosuprese. V následných cyklech je obvykle možné opět použít předchozí dávku, pokud se počty bílých krvinek a krevních destiček navrátily k přijatelným hodnotám.
Je-li v 1. dni jakéhokoli cyklu absolutní počet neutrofilů (ANC) < 1,0 x 109/l anebo je počet krevních destiček < 75 x 109/l, doporučuje se léčbu odložit, dokud se hodnota ANC nevrátí k > 1,0 x 109/l a počet krevních destiček k > 75 x 109/l.
Tabulky 1 a 2 jsou doporučeny jako vodítka k úpravám dávkování v 8. a 15. dni 28denních cyklů.
|
Tabulka 1 Úpravy dávkování kvůli hematologické toxicitě v 8. a 15. dni jakéhokoli cyklu | |||
|
Stupeň |
Počet krevních destiček |
Absolutní počet neutrofilů |
Úprava dávkování |
|
1-2 |
LLN* - 50 x 109/l |
LLN - 1,0 x 109/l |
Žádná změna dávky ani schématu léčby. |
|
3 |
< 50 - 25 x 109/l |
< 1,0 - 0,5 x 109/l |
Odložit léčbu, dokud se počet krevních destiček nevrátí na > 50 x 109/l a hodnota ANC** na > 1,0 x 109/l. |
|
4 |
< 25 x 109/l |
< 0,5 x 109/l |
Odložit léčbu, dokud se počet krevních destiček nevrátí na > 50 x 109/l a hodnota ANC** na > 1,0 x 109/l. Snížit dávku o 20 %. |
|
* LLN: Spodní limit normálního rozmezí (Lower Limit of the Normal range) ** ANC: Absolutní počet neutrofilů (Absolute Neutrophil Count) | |||
|
Tabulka 2 Úpravy léčby kvůli nehematologické toxicitě | |
|
Toxicita |
Úprava |
|
Jakákoli jiná než srdeční toxicita stupně 3 nebo 4 související s léčivem, kromě nauzey a zvracení |
Odložit léčbu, dokud se stav neupraví na stupeň 1. Snížit dávku o 20 %. |
|
Jakákoli kardiovaskulární toxicita NYHA* stupně 3 nebo 4 nebo přetrvávající pokles LVEF** |
Odložit léčbu a sledovat stav pacienta, dokud se neupraví. Zvážit přerušení léčby při přetrvávajícím poklesu LVEF** o hodnotě > 15 % výchozí hodnoty. |
|
* NYHA: New York Heart Association ** LVEF: Ejekční frakce levé komory (Left Ventricular Ejection Fraction) | |
Zvláštní skupiny pacientů Pediatrická populace
Bezpečnost a účinnost přípravku Pixuvri u dětí ve věku do 18 let nebyla dosud stanovena.
Nejsou dostupné žádné údaje.
Starší pacienti
U starších pacientů (ve věku od 65 let) není zapotřebí žádná zvláštní úprava dávkování.
Porucha funkce ledvin
Bezpečnost a účinnost přípravku Pixuvri u pacientů s poruchou funkce ledvin nebyla dosud stanovena. Pacienti s hodnotou sérového kreatininu > 1,5násobek horního limitu normálního rozmezí (Upper Limit of the Normal range (ULN)) byli z randomizované studie vyloučeni. Proto by se měl přípravek Pixuvri používat u pacientů s poruchou funkce ledvin obezřetně.
Pacienti s poruchou jaterních funkcí
Bezpečnost a účinnost přípravku Pixuvri u pacientů s poruchou jaterních funkcí nebyla dosud stanovena. Přípravek Pixuvri by se měl používat obezřetně u pacientů s mírnou nebo středně těžkou poruchou funkce jater. Přípravek Pixuvri se nedoporučuje používat u pacientů s těžkou poruchou exkreční funkce jater (viz bod 4.3).
Pacienti se špatným výkonnostním stavem
V současnosti nejsou k dispozici žádné informace o bezpečnosti a účinnosti u pacientů se špatným výkonnostním stavem (ECOG > 2). Při léčbě těchto pacientů by se mělo postupovat obezřetně.
Způsob podání
Přípravek Pixuvri je určen pouze k intravenóznímu podání. Bezpečnost intratekálního podání nebyla dosud stanovena.
Přípravek Pixuvri je určen k podání formou pomalé intravenózní infuze s použitím in-line filtru (po dobu nejméně 60 minut) pouze po rekonstituci v 5 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) a po dalším naředění injekčním roztokem chloridu sodného 9 mg/ml (0,9%) na konečný objem 250 ml.
Návod k rekonstituci a naředění tohoto léčivého přípravku před jeho podáním je uveden v bodě 6.6.
4.3 Kontraindikace
- Hypersenzitivita na pixantron-dimaleinát nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1
- Imunizace živými virovými vakcínami
- Závažná myelosuprese
- Těžká porucha funkce jater
4.4 Zvláštní upozornění a opatření pro použití
Veškeré úvodní léčbě přípravkem Pixuvri by mělo předcházet pečlivé výchozí vyšetření krevního obrazu, hladiny celkového bilirubinu v séru, hladiny celkového kreatininu v séru a srdeční funkce změřením ejekční frakce levé komory (LVEF).
Myelosuprese
Může se vyskytnout závažná myelosuprese. U pacientů léčených přípravkem Pixuvri existuje pravděpodobnost výskytu myelosuprese (neutropenie, leukopenie, anemie, trombocytopenie a lymfopenie), přičemž převažujícím projevem je neutropenie. Při doporučené dávce a schématu léčby je neutropenie obvykle přechodná, dosahuje nejnižších hodnot (nadiru) v období mezi 15.-22. dnem po podání léčiva v 1., 8. a 15. dni a k upravení stavu obvykle dojde do 28. dne.
Je nutné pečlivě sledovat krevní obraz, včetně počtu leukocytů, červených krvinek, krevních destiček a absolutního počtu neutrofilů. Je možné použít rekombinantní hematopoetické růstové faktory v souladu s instituciálními pokyny nebo pokyny Evropské společnosti pro lékařskou onkologii (European Society for Medical Oncology, ESMO). Měly by se zvážit úpravy dávkování (viz bod 4.2).
Kardiotoxicita
Během léčby přípravkem Pixuvri nebo po ní se mohou vyskytnout změny srdeční funkce, včetně snížení ejekční frakce levé komory nebo fatálního městnavého srdečního selhání.
Aktivní nebo skryté kardiovaskulární onemocnění, předchozí léčba antracykliny nebo antracendiony, předchozí nebo souběžná radioterapie aplikovaná na oblast mediastina nebo souběžné užívání jiných kardiotoxických léčivých přípravků může zvýšit riziko kardiotoxicity. Kardiotoxicita se může při používání přípravku Pixuvri objevit nezávisle na přítomnosti rizikových srdečních faktorů.
U pacientů se srdečním onemocněním nebo rizikovými faktory, jako jsou výchozí hodnota LVEF < 45 % při radionuklidové ventrikulografii (MUGA scan), klinicky významné kardiovaskulární
4
abnormality (odpovídající stupni 3 nebo 4 dle New York Heart Association, NYHA), infarkt myokardu v posledních 6 měsících, závažná arytmie, nekontrolovaná hypertenze, nekontrolovaná angina pectoris nebo předchozí kumulativní dávky doxorubicinu či jeho ekvivalentu přesahující 450 mg/m2, by mělo léčbě přípravkem Pixuvri předcházet pečlivé zvážení poměru přínosů a rizik.
Před zahájením léčby přípravkem Pixuvri, a poté pravidelně, by se měla sledovat srdeční funkce. Jestliže se během léčby projeví kardiotoxicita, musí se zvážit poměr přínosů a rizik pokračování v léčbě přípravkem Pixuvri.
Sekundární malignita
Rozvoj hematologických malignit, jako je sekundární akutní myeloidní leukémie (AML) nebo myelodysplastický syndrom (MDS), představuje známé riziko spojené s léčbou antracykliny nebo jinými inhibitory topoizomerázy II. K výskytu sekundárního nádorového onemocnění včetně AML a MDS může dojít během léčby přípravkem Pixuvri nebo po ní.
Infekce
V průběhu klinických studií byly hlášeny infekce, včetně pneumonie, celulitidy, bronchitidy a sepse (viz bod 4.8). Infekce byly spojeny s hospitalizací, septickým šokem a úmrtím. Pacienti s neutropenií jsou náchylnější k infekcím, přestože v klinických studiích nedošlo ke zvýšenému výskytu atypických, obtížně léčitelných infekcí, jako jsou systémové mykotické infekce či infekce oportunními organismy, např. Pneumocystis jiroveci.
Přípravek Pixuvri by se neměl podávat pacientům s aktivní závažnou infekcí ani pacientům s opakovanými či chronickými infekcemi v anamnéze nebo se základním onemocněním, které je může dále učinit náchylnými k závažné infekci.
Syndrom nádorového rozpadu
Pixantron může vyvolat hyperurikémii následkem rozsáhlého katabolismu purinů, který doprovází rychlý rozklad neoplastických buněk vyvolaný léčivem (syndrom rozpadu tumoru), a může vést k nerovnováze elektrolytů, jež může mít za následek poškození ledvin. U pacientů s vysokým rizikem rozpadu tumoru (zvýšení LDH, velký objem tumoru, vysoká výchozí hladina kyseliny močové nebo hladiny fosfátů v séru) by se měly po léčbě vyšetřit hladiny kyseliny močové, draslíku, fosforečnanu vápenatého a kreatininu v krvi. Hydratace, alkalizace moči a profylaxe alopurinolem nebo jinou látkou k zabránění hyperurikémie může minimalizovat možné komplikace syndromu nádorového rozpadu.
Imunizace
Imunizace může být neúčinná, je-li podána během léčby přípravkem Pixuvri. Imunizace živými virovými vakcínami je kontraindikována kvůli imunosupresi spojené s léčbou přípravkem Pixuvri (viz bod 4.3).
Extravazace
Dojde-li k extravazaci, mělo by se podávání přípravku okamžitě přerušit a přípravek by se měl znovu začít podávat do jiné žíly. Nepuchýřotvorné vlastnosti přípravku Pixuvri minimalizují riziko místní reakce po extravazaci.
Prevence fotosenzitivních reakcí
Fotosenzitivita představuje potenciální riziko na základě neklinických údajů in vitro a in vivo. V programu klinických studií nebyly hlášeny žádné potvrzené případy. Pacientům by se mělo jako preventivní opatření doporučit, aby dodržovali postupy ochrany před slunečním zářením, včetně nošení ochranného oblečení a používání ochranných kosmetických prostředků. Protože většina fotosenzitivních reakcí vyvolaných léčivými přípravky je způsobena vlnovými délkami v rozmezí UV-A, doporučují se ochranné kosmetické prostředky, které silně absorbují UV-A záření.
Pacienti na dietě s nízkým obsahem sodíku
Tento léčivý přípravek obsahuje po naředění přibližně 1000 mg (43 mmol) sodíku na jednu dávku.
To je třeba brát v úvahu při podávání tohoto léku pacientům, kteří drží dietu s nízkým obsahem sodíku.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
U lidí nebyly hlášeny žádné lékové interakce a nebyly provedeny žádné studie vzájemných lékových interakcí.
Studie inhibice in vitro
Studie in vitro s nejběžnějšími izoformami lidského cytochromu P450 (zahrnující CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6 a 3A4) ukázaly možnou inhibici smíšeného typu CYP1A2 a CYP2C8, která může mít klinický význam. Nebyly pozorovány žádné jiné klinicky významné interakce s izoformami CYPP450.
Theofylin: při současném podávání léčivého přípravku theofylinu s úzkým terapeutickým indexem, který je metabolizován především CYP1A2, existují teoretické obavy, že může dojít k zvýšení koncentrace tohoto substrátu vedoucí k toxicitě theofylinu. Hladina theofylinu by se měla pečlivě sledovat v týdnech následujících bezprostředně po zahájení souběžné léčby přípravkem Pixuvri.
Warfarin je částečně metabolizován pomocí CYP1A2, a proto existují teoretické obavy týkající se souběžného podávání tohoto léčivého přípravku a účinku, jaký může mít inhibice jeho metabolismu na jeho zamýšlené působení. Ve dnech následujících bezprostředně po zahájení souběžné léčby přípravkem Pixuvri by se měly sledovat koagulační parametry, konkrétně mezinárodní normalizovaný poměr (INR).
Amitriptylin, haloperidol, klozapin, ondansetron apropranololjsou metabolizovány pomocí CYP1A2, a proto existují teoretické obavy, že souběžné podávání přípravku Pixuvri může zvýšit hladiny tohoto léčivého přípravku v krvi.
Přestože riziko inhibice CYP2C8 pixantronem nebylo možné určit, je třeba postupovat obezřetně při souběžném podávání látek, které jsou metabolizovány především pomocí CYP2C8, jako repaglidinu, rosiglitazonunebopaklitaxelu, např. pečlivě sledovat vedlejší účinky.
Na základě studií in vitro se zjistilo, že pixantron je substrátem membránových transportních proteinů P-gp/BRCP a OCT1 a látky, které inhibují tyto transportéry, mají potenciální schopnost snížit vychytávání pixantronu játry a účinnost exkrece pixantronu. Při souběžném podávání látek, které inhibuj í tyto transportéry, jako j sou cyklosporin A nebo takrolimus běžně používané ke zvládnutí reakce štěpu proti hostiteli a přípravky proti HIV, jako jsou ritonavir, sachinavir nebo nelfinavir, by se měl pečlivě sledovat krevní obraz.
Dále by se mělo postupovat s obezřetností při kontinuálním podávání pixantronu souběžně s induktory efluxních transportérů, jako jsou rifampicin, karbamazepin a glukokortikoidy, neboť může dojít k zvýšenému vylučování pixantronu a následnému poklesu systémové expozice.
4.6 Fertilita, těhotenství a kojení
Ženy ve fertilním věku
Ženám ve fertilním věku a jejich partnerům by se mělo doporučit, aby se vyvarovali těhotenství.
Ženy a muži musí během léčby (a ještě 6 měsíců po ukončení terapie) používat účinnou antikoncepci.
Údaje o podávání pixantronu těhotným ženám nejsou k dispozici. Studie na zvířatech prokázaly reprodukční toxicitu (viz bod 5.3).
Podávání přípravku Pixuvri se v těhotenství a u žen v reprodukčním věku, které nepoužívají antikoncepci, nedoporučuje.
Kojení
Není známo, zda se přípravek Pixuvri/jeho metabolity vylučují do lidského mateřského mléka.
Riziko pro kojené novorozence /děti nelze vyloučit.
Kojení má být během léčby přípravkem Pixuvri přerušeno.
Fertilita
Po opakovaném podání přípravku Pixuvri v dávkách 0,1 mg/kg/den byla u psů zaznamenána atrofie varlat závislá na dávce. U lidí tento účinek nebyl dosud vyhodnocen. Stejně jako jiné látky z obecné třídy látek poškozujících deoxyribonukleovou kyselinu (DNK) může být přípravek Pixuvri spojen s poruchou fertility. Jelikož nebyl stanoven jeho účinek na fertilitu, je třeba pacientům mužského pohlaví v rámci bezpečnosti doporučit, aby používali během léčby a rovněž po dobu 6 měsíců po jejím ukončení antikoncepční metody (přednostně bariérové), což umožní dozrání nových spermií. Aby se zamezilo riziku dlouhodobé neplodnosti, mělo by se zvážit uchování spermatu ve spermabance.
4.7 Účinky na schopnost řídit a obsluhovat stroje
Není známo, zda přípravek Pixuvri má nějaký vliv na schopnost řídit nebo obsluhovat stroje.
4.8 Nežádoucí účinky Souhrn bezpečnostního profilu
Bezpečnost přípravku Pixuvri byla hodnocena u 407 pacientů.
Nejčastějším toxickým účinkem je myelosuprese, zejména linie neutrofilů. Přestože je výskyt myelosuprese s klinickými následky poměrně nízký, pacienti léčení přípravkem Pixuvri byli pečlivě sledováni pomocí častých vyšetření krevního obrazu, zejména s ohledem na neutropenii. Výskyt závažných infekcí byl nízký a oportunní infekce spojené s imunokompromitací nebyly pozorovány. Přestože výskyt kardiotoxicity projevující se městnavým selháním srdečním se zdá být nižší, než by se předpokládalo u příbuzných léčivých přípravků, např. antracyklinů, doporučuje se sledovat hodnotu LVEF buď pomocí MUGA scanů, nebo echokardiografického vyšetření (ECHO), aby se vyšetřila subklinická kardiotoxicita. Zkušenosti s pixantronem jsou omezeny na pacienty s hodnotou LVEF >
45 %, přičemž většina pacientů vykazovala hodnoty > 50 %. Zkušenosti s podáváním přípravku Pixuvri pacientům, kteří mají významněji oslabenou srdeční funkci, jsou omezené a podávání přípravku by se mělo provádět pouze v kontextu klinické studie. Další toxické účinky, jako jsou nauzea, zvracení a průjem, byly obecně nepříliš časté, mírné, vratné, zvládnutelné a u pacientů léčených cytotoxickými látkami očekávané. Účinky na jaterní nebo ledvinné funkce byly minimální, nebo se žádné nevyskytly.
Tabulkový souhrn nežádoucích účinků
Nežádoucí účinky hlášené v souvislosti s používáním přípravku Pixuvri pocházejí z konečných údajů všech dokončených studií. Nežádoucí účinky jsou uvedeny v tabulce 3 níže, seřazené podle tříd orgánových systémů a četnosti výskytu dle databáze MedDRA: velmi časté (>1/10), časté (>1/100 až <1/10), méně časté (>1/1000 až <1/100), vzácné (>1/10000 až <1/1000), velmi vzácné (<1/10000), není známo (z dostupných údajů nelze určit). V každé skupině četnosti jsou nežádoucí účinky seřazeny podle klesající závažnosti.
|
Nežádoucí účinky hláš< v dokončených studiích s |
Tabulka 3 ;né v souvislosti s přípravkem Pixuvri přípravkem Pixuvri seřazené dle četnosti | |
|
Třída orgánových systémů |
Frekvence |
Nežádoucí účinek |
|
Časté |
Neutropenická infekce, infekce dýchacích cest, infekce | |
|
Infekce a infestace |
Méně časté |
Bronchitida, kandidóza, celulitida, herpes zoster, meningitida, infekce nehtů, orální mykotická infekce, orální herpes, pneumonie, salmonella gastroenteritis, septický šok |
|
Novotvary benigní, maligní a blíže neurčené (zahrnující cysty a polypy) |
Méně časté |
Progrese novotvarů Sekundární malignita (zahrnující hlášení výskytu AML a MDS) |
|
Poruchy krve a lymfatického systému* |
Velmi časté |
Neutropenie, leukopenie, lymfopenie, anemie, trombocytopenie |
|
Časté |
Febrilní neutropenie, porucha krve | |
|
Méně časté |
Selhání kostní dřeně, eozinofilie | |
|
Poruchy imunitního systému |
Méně časté |
Přecitlivělost na léčivý přípravek |
|
Poruchy metabolismu a výživy |
Časté |
Anorexie, hypofosfatemie |
|
Méně časté |
Hyperurikemie, hypokalcemie, hyponatremie | |
|
Psychiatrické poruchy |
Méně časté |
Úzkost, nespavost, poruchy spánku |
|
Poruchy nervového systému |
Časté |
Poruchy vnímání chuti, parestézie, bolest hlavy, somnolence |
|
Méně časté |
Závratě, letargie | |
|
Poruchy oka |
Časté |
Konjunktivitida |
|
Méně časté |
Suché oko, keratitida | |
|
Poruchy ucha a labyrintu |
Méně časté | |
|
Srdeční poruchy* |
Časté |
Dysfunkce levé komory, srdeční porucha, městnavé selhání srdeční, raménkový blok, tachykardie |
|
Méně časté | ||
|
Cévní poruchy |
Časté |
Bledost, změna zabarvení žil, hypotenze |
|
Méně časté |
Žilní poruchy | |
|
Respirační, hrudní a |
Časté | |
|
mediastinální poruchy |
Méně časté |
Pleurální výpotek, pneumonitida, rinorea |
|
Velmi časté | ||
|
Gastrointestinální poruchy |
Časté |
Stomatitida, průjem, zácpa, bolest břicha, sucho v ústech, dyspepsie |
|
Méně časté |
Ezofagitida, orální parestézie, krvácení z rekta | |
|
Poruchy jater a žlučových cest |
Méně časté |
Hyperbilirubinémie |
|
Velmi časté |
Změna zabarvení kůže, alopecie | |
|
Poruchy kůže a podkožní tkáně* |
Časté |
Erytém, poruchy nehtů, svědění |
|
Méně časté | ||
|
Poruchy svalové a kosterní |
Časté |
Bolest kostí |
Nežádoucí účinky hlášené v souvislosti s přípravkem Pixuvri v dokončených studiích s přípravkem Pixuvri seřazené dle četnosti
|
Třída orgánových systémů |
F rekvence |
Nežádoucí účinek |
|
soustavy a pojivové tkáně |
Méně časté |
Bolest kloubů, artritida, bolest zad, svalová slabost, muskuloskeletální bolest hrudníku, muskuloskeletální ztuhlost, bolest šíje, bolesti v končetinách |
|
Poruchy ledvin a močových cest |
Velmi časté |
Chromaturie |
|
Časté |
Proteinurie, hematurie | |
|
Méně časté |
Oligurie | |
|
Poruchy reprodukčního systému a prsu |
Méně časté |
Spontánní erekce penisu |
|
Celkové poruchy a reakce v místě aplikace |
Velmi časté |
Astenie |
|
Časté |
Únava, zánět sliznic, horečka, bolest na hrudi, otok | |
|
Méně časté |
Zimnice, chlad v místě vpichu, lokální reakce | |
|
Vyšetření |
Časté |
Zvýšená hladina alaninaminotransferázy, zvýšená hladina aspartátaminotransferázy, zvýšená hladina alkalické fosfatázy v krvi, zvýšená hladina kreatininu v krvi |
|
Méně časté |
Bilirubin v moči, zvýšená hladina fosforu v krvi, zvýšená hladina močoviny v krvi, zvýšená hladina gama-glutamiltransferázy, zvýšený počet neutrofilů, snížení tělesné hmotnosti |
* Nežádoucí účinky podrobněji probrány níže
Tabulka 3
Popis vybraných nežádoucích účinků
Hematologické toxické účinky a komplikace neutropenie
Hematologické toxické účinky byly nej častěji se vyskytující pozorovanou toxicitou, avšak byly obecně snadno zvládnutelné pomocí imunostimulačních přípravků a podpůrné transfúze dle potřeby. Zatímco se v randomizované klinické studii vyskytovala neutropenie stupně 3-4 častěji u jedinců používajících přípravek Pixuvri, ve většině případů byla bez komplikací, nekumulativní a spojená s nízkým výskytem febrilní neutropenie nebo infekcí. Důležité však je, že obvykle nebyla zapotřebí podpora růstovým faktorem a transfúze červených krvinek a krevních destiček byly méně časté. (Viz bod 4.4)
Srdeční toxicita
Ve studii PIX 301 došlo u 13 pacientů (19,1 %) ve skupině s přípravkem Pixuvri k snížení ejekční frakce. U 11 pacientů léčených přípravkem Pixuvri byly tyto příhody stupně 1-2 a u 2 pacientů stupně 3; tyto příhody byly přechodné a nezávislé na dávce přípravku Pixuvri. Případy srdečního selhání (výrazy dle databáze MedDRA srdeční selhání a městnavé srdeční selhání) se vyskytly u 6 pacientů (8,8 %) léčených přípravkem Pixuvri (2 pacienti se stupněm 1-2, 1 pacient se stupněm 3 a 3 pacienti se stupněm 5). Tři pacienti léčení přípravkem Pixuvri (4,4 %) vykazovali tachykardii, arytmii, sinusovou tachykardii nebo bradykardii.
Doporučuje se provést výchozí vyšetření srdce pomocí MUGA scanu nebo vyšetření ECHO, zejména u pacientů s rizikovými faktory pro zvýšenou srdeční toxicitu. U pacientů s rizikovými faktory, jako je předchozí vysoká kumulativní expozice antracyklinům nebo významné, již existující srdeční onemocnění, by se mělo zvážit opakované stanovení ejekční frakce levé komory pomocí MUGA scanu nebo echokardiografie. (Viz bod 4.4)
Jiné časté toxické účinky
Změna zabarvení kůže a chromaturie jsou známé účinky související s podáváním přípravku Pixuvri, v důsledku zbarvení této sloučeniny (modré). Změna zabarvení kůže obvykle vymizí během několika dnů až týdnů, jakmile je léčivý přípravek vyloučen z těla.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
V souvislosti s přípravkem Pixuvri nebyly hlášeny žádné případy předávkování.
V klinických studiích se zvyšováním dávky se při podání jednotlivé dávky pixantronu až do množství 158 mg/m2 nevyskytly důkazy svědčící o toxicitě související s dávkou.
V případě předávkování se doporučuje podpůrná léčba.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Antineoplastické látky, antracykliny a příbuzné látky. ATC kód: L01DB11
Mechanismus účinku
Léčivou látkou přípravku Pixuvri je pixantron, cytotoxický aza-antracendion.
Na rozdíl od schválených antracyklinů (doxorubicin a jiné) a antracendionů (mitoxantron) je pixantron pouze slabý inhibitor topoizomerázy II. Navíc pixantron na rozdíl od antracyklinů či antracendionů přímo alkyluje DNA, čímž vytváří stabilní DNA adukty a dvouvláknové zlomy. Navíc, protože pixantron obsahuje ve struktuře kruhu heteroatom dusíku a nemá ketonové skupiny, vykazuje menší potenciální schopnost produkovat reaktivní formy kyslíku, vázat železo a vytvářet metabolity alkoholu, které se považují za příčinu kardiotoxicity antracyklinů. Díky této jedinečné struktuře vyvolal pixantron na zvířecích modelech minimální kardiotoxicitu v porovnání s doxorubicinem či mitoxantronem.
Souhrnná retrospektivní populační analýza FK/FD fáze 1 klinických studií a režimů kombinované léčby (fáze 1/2) prokázaly, že doba přežití bez progrese a neutropenie stupně 2-3 souvisely s expozicí přípravku Pixuvri.
Klinická účinnost a bezpečnost
Bezpečnost a účinnost přípravku Pixuvri jako monoterapie byla hodnocena v multicentrické, randomizované, aktivně kontrolované klinické studii u pacientů s relabujícím či refrakterním agresivním non-hodgkinským lymfomem, kteří již podstoupili nejméně dvě předchozí terapie (PIX301). V této studii bylo randomizováno 140 pacientů (1:1) do skupiny léčené buď přípravkem Pixuvri, nebo do srovnávací skupiny léčené chemoterapií s jednou látkou vybranou zkoušející osobou. Demografické údaje o pacientech a výchozí charakteristiky onemocnění byly mezi jednotlivými léčebnými skupinami dobře vyváženy a nebyly zaznamenány žádné statisticky významné rozdíly. Celkově činil medián věku pacientů ve studii 59 let, 61 % tvořili muži, 64 % byli běloši, 76 % mělo na začátku studie onemocnění stádia III/IV dle Ann Arbor, 74 % mělo výchozí skóre Mezinárodního prognostického indexu (IPI) > 2 a 60 % podstoupilo 3 a více předchozích chemoterapií. Pacienti s lymfomem z plášťových buněk nebyli do pivotní studie zařazeni. Požadovalo se, aby byli pacienti zařazení do studie PIX 301citliví na předchozí léčbu antracykliny (potvrzená nebo nepotvrzená kompletní odpověď (CR) nebo částečná odpověď (PR)).
Údaje u pacientů dříve léčených rituximabem (38 pacientů ve skupině s přípravkem Pixuvri a 39 pacientů ve skupině se srovnávacím přípravkem) jsou omezené .10
Odpověď nádoru na léčbu vyhodnotil zaslepený nezávislý centrální hodnotící výbor dle mezinárodního pracovního semináře ke standardizaci kritérií odpovědi u NHL. U pacientů léčených přípravkem Pixuvri byl významně vyšší výskyt kompletních odpovědí a nepotvrzených kompletních odpovědí (CR/CRu) a vyšší výskyt objektivní odpovědi (objective response rate, ORR) v porovnání se skupinou léčenou srovnávacím přípravkem (viz tabulku 4).
|
Tabulka 4 Souhrn odpovědí dle nezávislého hodnotícího výboru (populace ITT) | ||||||
|
Konec léčby |
Konec studie | |||||
|
Pixuvri (n=70) |
Srovnávací přípravek (n=70) |
p- hodnota |
Pixuvri (n=70) |
Srovnávací přípravek (n=70) |
p- hodnota | |
|
CR/CRu |
14 (20,0 %) |
4 (5,7 %) |
0,021 |
17 (24,3 %) |
5 (7,1 %) |
0,009 |
|
CR |
8 (11,4 %) |
0 (0 %) |
11 (15,7 %) |
0 (0,0 %) | ||
|
CRu |
6 (8,6 %) |
4 (5,7 %) |
6 (8,6 %) |
5 (7,1 %) | ||
|
ORR (CR, Cru a PR) |
26 (37,1 %) |
10 (14,3 %) |
0,003 |
28 (40,0 %) |
10 (14,3 %) |
0,001 |
|
K porovnání podílů ve skupině s přípravkem Pixuvri a skupině se srovnávacím c použit Fisherův exaktní test. |
íemoterapeutikem byl | |||||
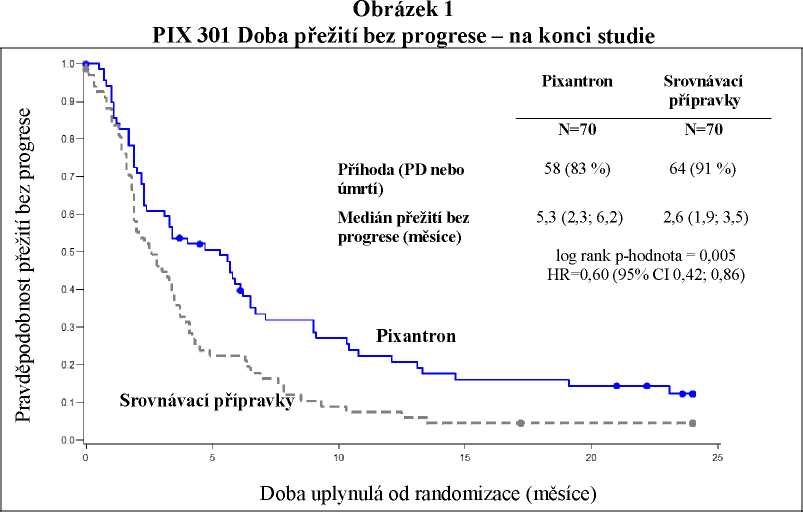
U pacientů léčených přípravkem Pixuvri došlo ke 40% prodloužení doby přežití bez progrese v porovnání s pacienty léčenými srovnávacími přípravky a medián doby přežití bez progrese byl o 2,7 měsíců delší (poměr rizik (HR)=0,60, logrank p=0,005) (viz níže obrázek 1).
Medián celkového přežití pacientů léčených přípravkem Pixuvri byl o 2,6 měsíců delší oproti pacientům léčeným srovnávacím přípravkem (HR=0,79, logrank p=0,25) (viz níže obrázek 2).
Obrázek 2
PIX 301 Celkové přežití - na konci studie
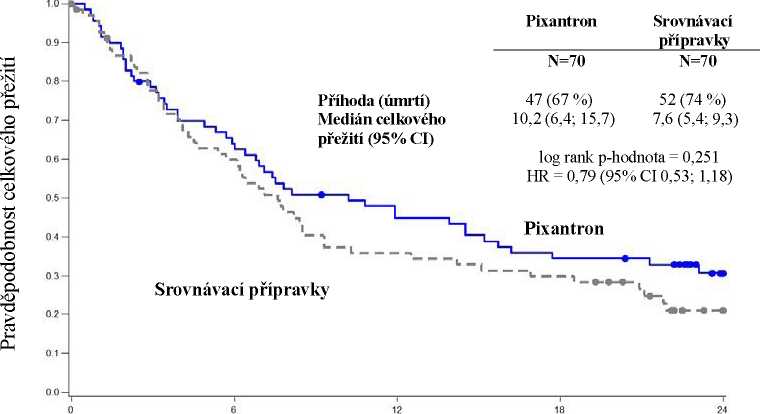
Doba uplynulá od randomizace (měsíce)
Výsledky u pacientů předléčených rituximabem nadále ukazují větší přínos léčby přípravkem Pixuvri oproti srovnávacímu přípravku s ohledem na výskyt celkové odpovědi na léčbu (31,6 % u přípravku Pixuvri oproti 17,9 % u srovnávacího přípravku) a medián přežití bez progrese (3,3 měsíce u přípravku Pixuvri oproti 2,5 měsíce u srovnávacího přípravku). Přínos přípravku Pixuvri však dosud nebyl stanoven při použití jako léčba páté a vyšší linie u pacientů nereagujících na poslední terapii a údaje u této skupiny pacientů jsou velmi omezené.
Pediatrická populace
Evropská agentura pro léčivé přípravky rozhodla o zproštění povinnosti předložit výsledky studií s přípravkem Pixuvri u kojenců od narození do věku 6 měsíců na základě skutečnosti, že se onemocnění NHL u této konkrétní pediatrické podskupiny nevyskytuje.
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Pixuvri u pacientů ve věku od 6 měsíců do 18 let s onemocněním NHL (informace o použití u dětí viz bod 4.2).
Tento léčivý přípravek byl registrován postupem tzv. podmíněného schválení. Znamená to, že jsou očekávány další důkazy o jeho přínosech.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tento souhrn údajů o přípravku bude podle potřeby aktualizován.
5.2 F arm akokinetické vlastnosti
Absorpce
Plazmatické koncentrace pixantronu dosáhly po intravenózním podání maximální koncentrace na konci infuze a poté polyexponenciálně klesaly. Farmakokinetika přípravku Pixuvri byla v rozmezí dávek 3 mg/m2 až 105 mg/m2 nezávislá na dávce a nebyly pozorovány žádné podstatné rozdíly při podávání tohoto léčivého přípravku samostatně nebo ve studiích kombinací přípravků. Průměrné expozice při samostatném podávání přípravku představovaly:
|
Dávka přípravku |
Počet pacientů |
AUC (0-24h) (ng.h/ml) |
|
Pixuvri (ma/m2) | ||
|
33 |
3 |
982± 115 |
|
49 |
6 |
1727 ±474 |
|
88 |
2 |
3811 |
Dle analýzy populačních FK údajů byl pro cílovou zaznamenanou dávku 50 mg/m2 pixantronu medián expozice v 28denním cyklu 6320 ng.h/ml (90% CI, 5990-6800 ng.h/ml), při 3 dávkách / 4týdenní cyklus.
Distribuce
Přípravek Pixuvri má velký distribuční objem 25,8 l a je přibližně z 50 % vázán na plazmatické proteiny.
Biotransformace
Acetylované metabolity jsou hlavními produkty biotransformace pixantronu. Nicméně za podmínek in vitro byla přeměna pixantronu na acetylované metabolity prostřednictvím NAT1 nebo NAT2 velmi omezená. Do lidské moči se látka vylučovala převážně nezměněná a byla nalezena pouze velmi malá množství acetylovaných metabolitů fáze I a fáze II. Proto se zdá, že metabolismus není důležitou cestou eliminace pixantronu. Acetylované metabolity byly farmakologicky neaktivní a metabolicky stabilní.
Eliminace
Pixantron má střední až vysokou celkovou plazmatickou clearance o hodnotě 72,7 l/h a nízkou renální exkreci odpovídající méně než 10 % podané dávky za 0-24 hodin. Terminální poločas se pohyboval v rozmezí od 14,5 do 44,8 h s průměrnou hodnotou 23,3 ± 8,0 (n=14, CV=34 %) a mediánem 21,2 h. Díky omezenému podílu renální clearance je plazmatická clearance převážně nerenální. Přípravek Pixuvri může být metabolizován v játrech nebo vylučován v žluči. Jelikož se metabolismus zdá být omezený, může být hlavní cestou eliminace vylučování nezměněného pixantronu žlučí. Jaterní clearance se blíží průtoku plazmy játry, což naznačuje vysoký extrakční poměr v játrech, a tudíž účinnou eliminaci mateřské léčivé látky. Vychytávání pixantronu játry se děje pravděpodobně prostřednictvím aktivních transportérů OTC1 a vylučování žlučí prostřednictvím transportérů P-gp a BCRP.
Pixantron měl pouze slabou nebo žádnou schopnost inhibovat transportní mechanismus P-gp, BCRP a BSEP in vitro.
Pixantron inhiboval in vitro transport metforminu prostřednictvím OTC1, avšak nepředpokládá se, že bude inhibovat OTC1 in vivo za klinicky relevantních podmínek.
Pixantron byl in vitro slabým inhibitorem transportérů j aterního vychytávání OATP1B1 a OATP1B3. Linearita / nelinearita
Farmakokinetika pixantronu se ukázala být lineární v širokém rozmezí dávek od 3 mg/m2 do 105
mg/m2.
Farmakokinetický(é)/farmakodynamický(é) vztah(y)
Byl pozorován vztah mezi plazmatickou expozicí pixantronu a počtem neutrofilů.
5.3 Předklinické údaje vztahující se k bezpečnosti
Po jednorázovém intravenózním podání přípravku Pixuvri v dávce 29 mg/kg a 38/mg/kg došlo u myší k okamžitým úmrtím (114 mg/m2, LD10). Byl pozorován pokles počtu bílých a červených krvinek a změny v kostní dřeni, slezině, ledvinách a varlatech. Podobné nálezy byly hlášeny u potkanů a u psů při dávce 116 mg/m2. U psů se bezprostředně po léčbě objevila tachykardie a změny na elektrokardiogramu (EKG).
Ve studiích s opakovanou dávkou u myší, potkanů a psů byly hlavními nálezy myelotoxicita, nefrotoxicita (s výjimkou psů) a poškození varlat.
U psů přípravek Pixuvri podaný v dávce 0,5 až 0,9 mg/kg v 6 cyklech nezpůsobil mortalitu ani závažné klinické příznaky, včetně změn EKG či tělesné hmotnosti. Samci byli na léčbu citlivější, pokud jde o snížení počtu bílých krvinek a krevních destiček a lymfoidní depleci (slezina a brzlík) a rovněž o výraznou toxicitu vůči reprodukčním orgánům, jak se u cytotoxické látky očekává. Kromě přechodného zvýšení expozice u samic po třetím cyklu nebyly pozorovány žádné výrazné rozdíly ve farmakokinetických parametrech. Samci však vykazovali nepatrně vyšší expozici než samice.
U psů neměla léčba vliv na srdce, neboť v různých obdobích léčby nebyly pozorovány žádné změny EKG, ani nebyly odhaleny žádné histopatologické či makropatologické změny na srdci. Také ledvinné funkce a histologický nález nebyly ve 4týdenní ani v 26týdenní studii ovlivněny.
Hodnotil se kardiotoxický potenciál přípravku Pixuvri v porovnání se stejně aktivními dávkami doxorubicinu a mitoxantronu v léčbě dosud neléčených myší a myší předléčených doxorubicinem. Pixantron-dimaleinát podávaný v dávce až 27 mg/kg dvakrát týdně po dobu 4 týdnů nevyvolal žádné kardiotoxické účinky, zatímco mitoxantron, jak se předpokládalo, byl ve všech testovaných dávkách (0,6, 1,6 a 1,5 mg/kg) kardiotoxický. Přípravek Pixuvri vyvolal mírnou nefropatii. Minimální kardiotoxicita přípravku Pixuvri byla rovněž prokázána v opakovaných léčebných cyklech se stejnými dávkami.
Studie genotoxicity potvrdily potenciální klastogenní účinky v savčích buňkách in vitro a in vivo.
V Amesově testu byl přípravek Pixuvri mutagenní, zvyšoval počet chromozomálních aberací v lidských lymfocytech a zvýšil frekvenci výskytu mikrojader in vivo.
Přípravek Pixuvri působil toxicky na matku a plod u potkanů a králíků, a to i v tak nízké dávce jako je dávka 1,8 mg/kg podaná 9.-11. den březosti, přičemž vyšší dávky vedly k potratům a úplné resorpci embrya. Embryotoxicita byla charakterizována snížením průměrné hmotnosti plodu, malformacemi plodu a nedokončenou nebo opožděnou fetální osifikací. Nebyly provedeny žádné dlouhodobé studie na zvířatech ke stanovení karcinogenního potenciálu přípravku Pixuvri. Nebyla provedena žádná studie lokální snášenlivosti.
Bylo prokázáno, že přípravek Pixuvri má in vitro fototoxické účinky na 3T3 buňky.
Ve studii jednotek tvořících kolonii u myší byla myelotoxicita přípravku Pixuvri a mitoxantronu podávaných v dávce LD10 (pixantron-dimaleinát 38 mg/kg a mitoxantron 6,1 mg/kg) podobná.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Chlorid sodný Monohydrát laktózy Hydroxid sodný (k úpravě pH)
Kyselina chlorovodíková (k úpravě pH)
6.2 Inkompatibility
Tento léčivý přípravek nesmí být mísen s jinými léčivými přípravky s výjimkou těch, které jsou uvedeny v bodě 6.6.
6.3 Doba použitelnosti
Neotevřená injekční lahvička 5 let
Roztok po rekonstituci a naředění
Chemická a fyzikální stabilita po otevření před použitím byla prokázána na dobu 24 hodin při pokojové teplotě (15°C až 25°C) a vystavení dennímu světlu ve standardních polyethylenových (PE) infuzních vacích.
Z mikrobiologického hlediska má být přípravek použit okamžitě. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2°C až 8°C, pokud rekonstituce a ředění neproběhly za kontrolovaných a validovaných aseptických podmínek.
6.4 Zvláštní opatření pro uchovávání
Uchovávejte v chladničce (2°C - 8°C).
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Podmínky uchovávání po rekonstituci a naředění léčivého přípravku viz bod 6.3.
6.5 Druh obalu a obsah balení
Skleněná inj ekční lahvička typu I s šedou butylpryžovou zátkou a hliníkovou pertlí a červeným plastovým víčkem obsahující 50 mg pixantron-dimaleinátu, což odpovídá 29 mg pixantronu.
Velikost balení po 1 lahvičce.
6.6 Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Rekonstituce a naředění
Před rekonstitucí lyofilizovaný prášek zkontrolujte zrakem, zda nevykazuje neobvyklé vady, např. trhliny, roztavení nebo sklovitý vzhled. Asepticky přidejte do každé 29 mg injekční lahvičky 5 ml injekčního roztoku chloridu sodného o koncentraci 9 mg/ml (0,9%). Lyofilizovaný prášek by se měl za protřepávání zcela rozpustit během 60 sekund. Vznikne tmavě modrý roztok o koncentraci pixantronu 5,8 mg/ml.
Asepticky natáhněte potřebný objem pro požadovanou dávku (vycházející z koncentrace 5,8 mg/ml) a vstříkněte jej do infuzního vaku o objemu 250 ml obsahujícího injekční roztok chloridu sodného o koncentraci 9 mg/ml (0,9%). Konečná koncentrace pixantronu v infuzním vaku by měla být nižší než 580 mikrogramů/ml na základě vstupního množství rekonstituovaného léčivého přípravku. Kompatibilita s jinými ředicími roztoky nebyla dosud stanovena. Po přidání přípravku do infuzního vaku důkladně promíchejte obsah vaku. Výslednou směsí by měl být čirý, tmavě modrý roztok.
Při podávání naředěného roztoku přípravku Pixuvri by se měly používat in-line filtry o velikosti pórů 0,2 pm z polyethersulfonu.
Přípravek Pixuvri je cytotoxická látka. Zamezte kontaktu přípravku s očima nebo pokožkou. Při manipulaci s přípravkem Pixuvri a během dekontaminačních postupů používejte rukavice, masky a pomůcky na ochranu očí.
Zvláštní opatření pro likvidaci přípravku
Přípravek Pixuvri je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad včetně materiálu použitého k rekonstituci, naředění a podání přípravku musí být zlikvidován v souladu s místními požadavky platnými pro cytotoxické látky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT Velká Británie
8. REGISTRAČNÍ ČÍSLO (A)
EU/1/12/764/001
9. DATUM PRVNÍ REGISTRACE/PRODLOUŽENÍ REGISTRACE
Datum první registrace: 10. května 2012
Datum posledního prodloužení registrace: 22. března 2016
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Název a adresa výrobce odpovědného za propouštění šarží
Catalent UK Packaging Limited Lancaster Way, Wingates Industrial Estate Westhoughton, Bolton Lancashire BL5 3XX Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
• Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
E. ZVLÁŠTNÍ POVINNOST USKUTEČNIT POREGISTRAČNÍ OPATŘENÍ PRO PODMÍNĚNOU REGISTRACI PŘÍPRAVKU
Tato registrace byla schválena postupem tzv. podmíněného schválení, a proto podle článku 14(7) nařízení (ES) č. 726/2004 držitel rozhodnutí o registraci uskuteční v daném termínu následující opatření:
|
Popis |
Termín splnění |
|
Provedení randomizované kontrolované studie fáze 3 (PIX306) pixantron- rituximab versus gemcitabin-rituximab u pacientů s agresivním NHL z B-buněk, u kterých selhala léčba první linie CHOP-R a kteří nej sou způsobilí k autologní transplantaci kmenových buněk (ASCT) (2. linie) nebo u nich tato transplantace selhala (3. nebo 4. linie). Má být předložena zpráva o klinické studii. |
31. prosince 2018 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Pixuvri 29 mg prášek pro koncentrát pro infuzní roztok pixantronum
Jedna injekční lahvička obsahuje pixantron-dimaleinát v množství, které odpovídá 29 mg pixantronu. Po rekonstituci jeden ml koncentrátu obsahuje pixantron-dimaleinát v množství, které odpovídá 5,8 mg pixantronu.
Monohydrát laktózy, chlorid sodný, kyselina chlorovodíková, hydroxid sodný. Obsahuje sodík; další informace naleznete v příbalové informaci.
Prášek pro koncentrát pro infuzní roztok. Velikost balení 1 injekční lahvička.
Před použitím proveďte rekonstituci a naředění přípravku. Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Cytotoxická látka: Manipulujte s ní opatrně.
EXP
Uchovávejte v chladničce (2°C - 8°C).
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
m ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT Velká Británie
č.s.:
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato.
Pixuvri 29 mg prášek pro koncentrát pro infuzní roztok pixantronum
Jedna injekční lahvička obsahuje pixantron-dimaleinát v množství, které odpovídá 29 mg pixantronu. Po rekonstituci jeden ml koncentrátu obsahuje pixantron-dimaleinát, v množství, které odpovídá 5,8 mg pixantronu.
Monohydrát laktózy, chlorid sodný, kyselina chlorovodíková, hydroxid sodný. Obsahuje sodík; další informace najdete v příbalové informaci.
Prášek pro koncentrát pro infuzní roztok.
Před použitím proveďte rekonstituci a naředění přípravku. Před použitím si přečtěte příbalovou informaci. Intravenózní podání.
Uchovávejte mimo dohled a dosah dětí.
Cytotoxická látka: Manipulujte s ní opatrně.
EXP
Uchovávejte v chladničce (2°C - 8°C).
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Veškerý nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT Velká Británie
EU/1/12/764/001
č.š.:
Výdej léčivého přípravku vázán na lékařský předpis.
Nevyžaduje se - odůvodnění přijato.
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro uživatele
Pixuvri 29 mg prášek pro koncentrát pro infuzní roztok
Pixantronum
VTento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek používat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Pixuvri a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Pixuvri používat
3. Jak se přípravek Pixuvri používá
4. Možné nežádoucí účinky
5. Jak přípravek Pixuvri uchovávat
6. Obsah balení a další informace
1. Co je přípravek Pixuvri a k čemu se používá
Přípravek Pixuvri patří do farmakoterapeutické skupiny léčiv zvané „antineoplastické látky“. Ty se používají k léčbě rakoviny.
Přípravek Pixuvri se používá k léčbě dospělých pacientů s opakovaně relabujícími nebo refrakterními agresivními non-hodgkinskými lymfomy. Přípravek Pixuvri zabíjí rakovinné buňky tím, že se naváže na jejich DNA, což vede k usmrcení těchto buněk. Používá se u pacientů, jejichž rakovina nereaguje na léčbu nebo se po léčbě jinými chemoterapeutiky znovu vrátila.
2. Čemu musíte věnovat pozornost, než začnete přípravek Pixuvri používat Nepoužívejte přípravek Pixuvri:
- jestliže j ste alergický(á) na pixantron-dimaleinát nebo kteroukoli další složku tohoto přípravku
(uvedenou v bodě 6).
- jestliže Vám byla v nedávné době podána vakcína.
- jestliže Vám bylo sděleno, že trpíte přetrvávajícím dlouhodobě nízkým počtem červených krvinek, bílých krvinek a krevních destiček.
- jestliže trpíte velmi závažnými jaterními potížemi.
Upozornění a opatření
Poraďte se se svým lékařem, než začnete používat přípravek Pixuvri:
- jestliže Vám bylo sděleno, že máte velmi nízký počet bílých krvinek,
- jestliže trpíte srdečním onemocněním nebo nekontrolovaným vysokým krevním tlakem, zej ména pokud Vám již bylo někdy sděleno, že j ste měl(a) srdeční selhání, nebo pokud jste měl(a) v posledních 6 měsících srdeční infarkt,
- jestliže máte infekci,
jestliže jste již někdy podstoupil(a) léčbu rakoviny, jestliže dodržujete zvláštní dietu s omezeným příjmem sodíku,
jestliže užíváte další léčivé přípravky, které by se mohly s přípravkem Pixuvri navzájem ovlivňovat (viz „Další léčivé přípravky” níže).
Citlivost pokožky vůči slunečnímu záření
Během léčby pixantronem byste měl(a) minimalizovat nebo se vyhnout přírodnímu a umělým zdrojům slunečního záření (solária nebo léčba zářením UVA/B). Pokud budete vystaven(a) slunečnímu záření, měl(a) byste nosit ochranný oděv a používat ochranné kosmetické prostředky, které silně absorbují záření UV-A.
Děti a dospívající
Nepodávejte tento léčivý přípravek dětem mladším 18 let, neboť nejsou k dispozici žádné informace o léčbě přípravkem Pixuvri u dětí a dospívajících.
Další léčivé přípravky a přípravek Pixuvri
Informujte svého lékaře o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat. Je to nesmírně důležité, neboť užíváním několika různých léků zároveň se může účinek těchto léků
zeslabovat nebo zesilovat. Proto nesmíte přípravek Pixuvri používat spolu s dalšími léky, pokud Váš lékař nepotvrdil, že je to bezpečné.
Rozhodně svého lékaře informujte, užíváte-li nebo jste v nedávné době užíval(a) kterýkoli z následujících léků:
Užíváte-li následující léky, sdělte to svému lékaři:
- warfarin k prevenci tvorby krevních sraženin,
- theofylin k léčbě plicních onemocnění jako emfyzém nebo astma,
- amitriptylin k léčbě deprese,
- olanzapin, klozapin k léčbě schizofrenie nebo maniakální deprese,
- haloperidol k léčbě úzkosti a nespavosti,
- ondansetron k prevenci pocitu na zvracení a zvracení během chemoterapie,
- propanolol k léčbě vysokého krevního tlaku.
Přípravek Pixuvri s jídlem a pitím
Své stravovací návyky nemusíte po léčbě přípravkem Pixuvri měnit, pokud Vám to Váš lékař nenařídil.
Těhotenství, kojení a plodnost
Přípravek Pixuvri se nesmí podávat těhotným ženám, neboť může způsobit poškození nenarozeného dítěte. Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem nebo lékárníkem dříve, než začnete tento přípravek používat.
Během používání přípravku Pixuvri a prvních 6 měsíců po ukončení léčby se musí používat vhodná antikoncepční opatření. Týká se to žen, které mohou otěhotnět, a mužů používajících přípravek Pixuvri, kteří jsou schopni zplodit dítě.
Během léčby přípravkem Pixuvri nekojte.
Řízení dopravních prostředků a obsluha strojů
Není známo, zda přípravek Pixuvri ovlivňuje schopnost řídit nebo obsluhovat stroje.
Informace pro pacienty na dietě s nízkým obsahem soli
Tento léčivý přípravek obsahuje po naředění přibližně 1000 mg (43 mmol) sodíku na jednu dávku. To je třeba brát v úvahu při podávání tohoto léku pacientům, kteří drží dietu s nízkým obsahem sodíku.
Jak se přípravek Pixuvri používá
3.
Jaké množství přípravku Pixuvri se podává
Množství (dávka) přípravku Pixuvri, které Vám bude podáno, závisí na ploše povrchu Vašeho těla v metrech čtverečních (m2). To se určí z Vaší výšky a tělesné hmotnosti. V úvahu se rovněž vezmou výsledky krevních testů a Váš zdravotní stav. Doporučená dávka je 50 mg/m2. Je-li to třeba, Váš lékař dávku během léčby upraví.
Předtím, než Vám bude přípravek Pixuvri podán, Vám Váš lékař provede určité testy.
Jak často se přípravek Pixuvri používá
Přípravek Pixuvri se podává 1., 8. a 15. den každého 28denního cyklu po dobu až 6 cyklů.
Před podáním infuze možná dostanete léky k prevenci a omezení možných reakcí na přípravek Pixuvri, například léky k prevenci nevolnosti.
Jak se přípravek Pixuvri podává
Přípravek Pixuvri se podává kapačkou do žíly (intravenózní infuzí). Tento úkon provede zdravotní sestra nebo lékař.
Jak dlouho bude infuze trvat
Infuze Vám bude podávána po dobu přibližně jedné hodiny, není-li určeno jinak.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Reakce na infuzi
Během podávání infuze přípravku Pixuvri se může vzácně vyskytnout bolest/zarudnutí v místě vpichu. Pokud cítíte bolest nebo místo vpichu zrudne, okamžitě to sdělte osobě, která Vám infuzi podává. Může být nutné infuzi zpomalit nebo přerušit. Poté, co tyto příznaky odezní nebo se zmírní, může se v infuzi pokračovat.
Přípravek Pixuvri má tmavě modrou barvu, a proto se u Vás po několik dní po jeho podání může rozvinout namodralé zabarvení kůže a očí a Vaše moč může mít namodralou barvu. Zabarvení kůže obvykle vymizí během několika dnů až týdnů, jakmile je léčivý přípravek vyloučen z těla.
Infekční onemocnění
Jestliže se u Vás po léčbě přípravkem Pixuvri objeví příznaky infekce (například horečka, zimnice, namáhavé dýchání, kašel, vředy v ústech, potíže s polykáním nebo závažný průjem), sdělte to svému lékaři. Poté, co Vám byl podán přípravek Pixuvri, můžete být náchylnější k infekcím.
Srdce
Existuje možnost, že by se následkem léčby snížila schopnost Vašeho srdce pumpovat krev, nebo se u Vás může dokonce rozvinout závažný stav zvaný srdeční selhání, zejména pokud máte oslabenou funkci srdce již na začátku léčby přípravkem Pixuvri. Váš lékař bude sledovat Vaše srdeční funkce, zda se neobjevují příznaky či symptomy postižení srdce.
Pokud se domníváte, že trpíte kteroukoli z následujících reakcí, sdělte to svému lékaři
Velmi časté nežádoucí účinky (mohou postihnout více než 1 osobu z 10):
- zabarvení pokožky
- prořídnutí nebo vypadání vlasů
- abnormální zabarvení moči
- fyzická slabost
- nízký počet bílých krvinek, nízký počet červených krvinek (anémie) a nízký počet krevních destiček v krvi (může být nutná transfúze).
Časté nežádoucí účinky (mohou postihnout více než 1 osobu ze 100):
- infekce, např. infekce plic, kožní infekce, infekce spojené s nízkým počtem bílých krvinek, kvasinková infekce
- horečka
- poruchy vnímání chuti
- abnormální pocity na kůži, jako je pocit necitlivosti, brnění, píchání (parestézie)
- bolest hlavy
- ospalost
- únava
- zánět očí (konjunktivitida)
- průjem
- bolesti břicha
- zánět a/nebo vředy v krku a ústech
- sucho v ústech, zácpa, zažívací potíže, nechutenství
- změny na kůži j ako zarudnutí a svědění kůže, změny nehtů
- poškození srdce, snížení schopnosti srdce pumpovat krev, blokáda elektrických signálů v srdci, nepravidelný nebo rychlý srdeční rytmus
- nízký krevní tlak
- změna zabarvení žil, bledá pokožka
- dušnost, kašel
- krev v moči
- nadměrné množství bílkovin v moči
- otoky nohou nebo kotníků či j iných částí těla
- bolest kostí
- bolest na hrudi
- nízké hladiny fosfátů v krvi
- abnormální výsledky jaterních krevních testů nebo ledvinových funkcí.
Méně časté nežádoucí účinky (mohou postihnout více než 1 osobu z 1000):
- závažné infekce, jako je septický šok, bronchitida, pneumonie, kandidóza, celulitida, meningitida, gastroenteritida
- virové infekce, jako je pásový opar, nebo reaktivace jiného viru, např. herpes v ústech
- nervozita, nespavost
- ztráta energie
- točení hlavy, závratě
- suchost oka
- necitlivost v ústech
- infekce rohovky
- alergie na léčivý přípravek
- snížení obsahu vápníku a sodíku v krvi, zvýšení hladiny kyseliny močové v krvi
- zánět nebo nahromadění tekutiny okolo plic
- vodnatá rýma
- krvácení, např. střevní krvácení, červené skvrnky po těle v důsledku popraskání krevních cév
- podráždění žíly
- noční pocení
- nepravidelný srdeční rytmus
- spontánní erekce
- kožní vyrážka a/nebo vředy
- bolest, otoky, slabost a ztuhlost kloubů nebo svalů
- snížený výdej moči
- ztráta tělesné hmotnosti
- zvýšená hladina bilirubinu v krvi nebo moči
- zánět jícnu
bolest šíje, zad a končetin infekce nehtů
progrese novotvarů (tumorů)
nově vzniklý zhoubný nádor kostní dřeně nebo krve, jako je akutní myeloidní leukémie
(AML) nebo myelodysplastický syndrom (MDS)
selhání kostní dřeně
zvýšený počet eozinofilů v krvi.
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
5. Jak přípravek Pixuvri uchovávat
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte přípravek po uplynutí doby použitelnosti uvedené na štítku injekční lahvičky a na krabičce za zkratkou „EXP“. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte v chladničce (2°C - 8°C).
Uchovávejte injekční lahvičku v krabičce, aby byl přípravek chráněn před světlem.
Přípravek Pixuvri neobsahuje žádnou složku zabraňující růstu bakterií, a proto se doporučuje použít jej bezprostředně po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2°C - 8°C.
Roztok pixantronu je po rekonstituci stabilní po 24 hodin při pokojové teplotě (15°C až 25 °C) ve standardních infuzních vacích.
Přípravek Pixuvri je určen pouze k jednorázovému použití. Veškerý nepoužitý léčivý přípravek nebo odpad včetně materiálu použitého k rekonstituci, naředění a podání přípravku musí být zlikvidován v souladu s místními požadavky.
6. Obsah balení a další informace Co přípravek Pixuvri obsahuje
- Léčivou látkou je pixantronum. Jedna injekční lahvička obsahuje 50 mg pixantron-
dimaleinátu (to odpovídá 29 mg pixantronu). Dalšími složkami jsou monohydrát laktózy, hydroxid sodný, kyselina chlorovodíková a chlorid sodný.
Jak přípravek Pixuvri vypadá a co obsahuje toto balení
Přípravek Pixuvri je prášek pro přípravu koncentrátu pro přípravu infuzního roztoku. Je to tmavě modrý prášek, který se dodává v injekčních lahvičkách obsahujících 29 mg pixantronu. Velikost balení: 1 injekční lahvička.
Držitel rozhodnutí o registraci
CTI Life Sciences Limited Highlands House Basingstoke Road Spencers Wood, Reading Berkshire RG7 1NT
Velká Británie
Výrobce
Catalent UK Packaging Limited Lancaster Way, Wingates Industrial Estate Westhoughton, Bolton,
Lancashire BL5 3XX Velká Británie
Další informace o tomto přípravku získáte u místního zástupce držitele rozhodnutí o registraci
|
Belgie/Belgique/Belgien S.A. Servier Benelux N.V. Tél/Tel: +32 (0)2 529 43 11 |
Lietuva UAB “SERVIER PHARMA” Tel: +370 (5) 2 63 86 28 |
|
Etarapna CepBne MegHKaa EOOfl Tea.: +359 2 921 57 00 |
Luxembourg/Luxemburg S.A. Servier Benelux N.V. Tel: +32 (0)2 529 43 11 |
|
Česká republika Servier s.r.o. Tel: +420 222 118 111 |
Magyarország Servier Hungaria Kft. Tel: +36 1 238 7799 |
|
Danmark CTI Life Sciences Limited Tlf: + 45 36 92 77 96 |
Malta GALEPHARMA Ltd Tel: +(356) 21 247 082 |
|
Deutschland CTI Life Sciences Limited Tel.: + 49 (0)6922 223384 |
Nederland Servier Nederland Farma B.V. Tel: + 31 (0)71 5246700 |
|
Eesti Servier Laboratories OU Tel:+ 372 664 5040 |
Norge CTI Life Sciences Limited Tlf: + 47 21 03 39 98 |
|
EkXába IEPBIE EAAAI OAPMAKEYTIKH EnE Tr|U + 30 210 939 1000 |
Osterreich CTI Life Sciences Limited Tel: + 43 (0)19 287 896 |
|
Espaňa Laboratorios Servier S.L. Tel: + 34 91 748 96 30 |
Polska Servier Polska Sp. z o.o. Tel: + 48 (0) 22 594 90 00 |
|
France Les Laboratoires Servier Tél: + 33 (0)1 55 72 60 00 |
Portugal Servier Portugal, Lda Tel: + 351 21 312 20 00 |
|
Hrvatska Servier Pharma, d. o. o. Tel: + 385 (0)1 3016 222 |
Románia Servier Pharma SRL Tel: + 4 021 528 52 80 |
|
Ireland Servier Laboratories (Ireland) Ltd. Tel: + 353 (0)1 663 8110 |
Slovenija Servier Pharma d. o. o. Tel: + 386 (0)1 563 48 11 |
|
Island Servier Laboratories |
Slovenská republika Servier Slovensko spol. s r.o. |
c/o Icepharma hf Tel: + 421 2 5920 41 11
Sími: + 354 540 8000
Italia
Servier Italia S.p.A. Tel: + 39 (06) 669081
Kúrcpog
CA Papaellinas Ltd. T^k: + 357 22 741 741
Latvija
SIA Servier Latvia Tel: + 371 67502039
Suomi/Finland
CTI Life Sciences Limited Puh/Tel: + 358 9 23 195 429
Sverige
CTI Life Sciences Limited Tel: + 46 (0) 850 33 48 19
United Kingdom
CTI Life Sciences Limited Tel: + 44 (0)800 083 4014
Tato příbalová informace byla naposledy revidována
Tomuto léčivému přípravku bylo uděleno tzv. podmíněné schválení. Znamená to, že informace o tomto přípravku budou přibývat.
Evropská agentura pro léčivé přípravky nejméně jednou za rok vyhodnotí nové informace o tomto léčivém přípravku a tato příbalová informace bude podle potřeby aktualizována.
Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
Na webových stránkách Evropské agentury pro léčivé přípravky je tato příbalová informace k dispozici ve všech úředních jazycích EU/EHP.
Následující informace jsou určeny pouze pro zdravotnické pracovníky:
Podrobné nokvnv pro uživatele
PŘED REKONSTITUCÍ SI PŘEČTĚTE CELÉ POKYNY K PŘÍPRAVĚ Zvláštní opatření pro použití
Přípravek Pixuvri je protinádorový léčivý přípravek, který poškozuje buňky, a proto je třeba při manipulaci s ním postupovat opatrně. Zamezte kontaktu přípravku s očima nebo pokožkou. Při manipulaci s přípravkem a během dekontaminačních postupů používejte rukavice, masky a pomůcky na ochranu očí. Pokud se přípravek Pixuvri (lyofilizovaný prášek nebo tekutý roztok po rekonstituci) dostane do styku s pokožkou, ihned pokožku omyjte a důkladně membrány opláchněte vodou.
Rekonstituce/příprava pro intravenózní podání
Jedna injekční lahvička přípravku Pixuvri určená k jednomu použití obsahuje pixantron-dimaleinát v množství odpovídajícím 29 mg pixantronu. Po rekonstituci pomocí 5 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%) jeden ml koncentrátu obsahuje pixantron-dimaleinát, v množství, které odpovídá 5,8 mg pixantronu.
Před rekonstitucí zkontrolujte zrakem lyofilizovaný prášek, zda nevykazuje neobvyklé vady, např. trhliny, roztavení nebo sklovitý vzhled. Sterilním postupem rekonstituujte obsah každé 29mg injekční lahvičky s 5 ml injekčního roztoku chloridu sodného 9 mg/ml (0,9%). Prášek by se měl za protřepávání zcela rozpustit během 60 sekund. Vznikne tak tmavě modrý roztok o koncentraci pixantronu 5,8 mg/ml.
Sterilním postupem natáhněte objem potřebný pro požadovanou dávku (na základě koncentrace 5,8 mg/ml) a vstříkněte jej do 250ml infuzního vaku s injekčním roztokem chloridu sodného 9 mg/ml (0,9%). Kompatibilita s jinými ředicími roztoky nebyla dosud stanovena. Po přidání přípravku do infuzního vaku důkladně promíchejte obsah vaku. Výslednou směsí by měl být tmavě modrý roztok.
Při podávání naředěného roztoku přípravku Pixuvri by se měly používat in-line filtry s póry o velikosti 0,2 pm z polyethersulfonu.
Podmínky uchovávání po rekonstituci
Přípravek Pixuvri neobsahuje žádnou složku zabraňující růstu bakterií, a proto se doporučuje použít jej bezprostředně po rekonstituci. Není-li použit okamžitě, doba a podmínky uchovávání přípravku po otevření před použitím jsou v odpovědnosti uživatele a normálně by doba neměla být delší než 24 hodin při 2°C - 8°C.
Roztok je po rekonstituci a naředění stabilní po 24 hodin při pokojové teplotě (15°C až 25 °C) při vystavení dennímu světlu ve standardních polyethylenových (PE) infuzních vacích.
Zvláštní opatření pro likvidaci přípravku a pro zacházení s ním
Přípravek Pixuvri je cytotoxická látka. Všechen nepoužitý přípravek nebo odpad musí být zlikvidován v souladu s místními požadavky.
Přístroje a povrchy náhodně kontaminované přípravkem Pixuvri se musí ošetřit roztokem chlornanu sodného (100 pl vody a 20 pl chlornanu sodného [7 ± 2 % dostupného chloru] na 0,58 mg přípravku Pixuvri).
S vybavením, jako jsou injekční lahvičky, jehly a stříkačky použité při podávání přípravku Pixuvri, by se mělo zacházet jako s toxickým odpadem.
34