Orkambi 200 Mg/125 Mg
PŘÍLOHA I
SOUHRN ÚDAJŮ O PŘÍPRAVKU
Tento léčivý přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Žádáme zdravotnické pracovníky, aby hlásili jakákoli podezření na nežádoucí účinky. Podrobnosti o hlášení nežádoucích účinků viz bod 4.8.
1. NÁZEV PŘÍPRAVKU
Orkambi 200 mg/125 mg potahované tablety
2. KVALITATIVNÍ A KVANTITATIVNÍ SLOŽENÍ
Jedna potahovaná tableta obsahuje lumacaftorum 200 mg a ivacaftorum 125 mg. Úplný seznam pomocných látek viz bod 6.1.
3. LÉKOVÁ FORMA
Potahovaná tableta (tableta)
Růžové tablety oválného tvaru (o rozměrech 14 x 8,4 x 6,8 mm) s černě vytištěným označením „2V125“ na jedné straně.
4. KLINICKÉ ÚDAJE
4.1 Terapeutické indikace
Přípravek Orkambi je indikován k léčbě cystické fibrózy (CF) u pacientů ve věku od 12 let, kteří jsou homozygotními nosiči mutace F508del v genu CFTR (viz body 4.4 a 5.1).
4.2 Dávkování a způsob podání
Přípravek Orkambi smí předepisovat pouze lékaři, kteří mají zkušenosti s léčbou CF. Pokud je genotyp pacienta neznámý, má být přítomnost mutace F508del na obou alelách genu CFTR potvrzena přesnou a validovanou metodou genotypování.
Dávkování
Doporučená dávka je dvě tablety (jedna tableta obsahuje 200 mg lumakaftoru / 125 mg ivakaftoru) užívané perorálně každých 12 hodin (celková denní dávka 800 mg lumakaftoru / 500 mg ivakaftoru).
Přípravek Orkambi se má užívat s jídlem obsahujícím tuk. Jídlo nebo svačina obsahující tuk se má sníst přímo před užitím dávky nebo ihned po jejím užití. (viz bod 5.2).
Vynechaná dávka
Pokud od doby, kdy pacient dávku obvykle užívá, uplynulo méně než 6 hodin, má pacient plánovanou dávku přípravku Orkambi užít ihned s jídlem obsahujícím tuk. Pokud uplynulo více než 6 hodin, má pacient počkat a užít až následující plánovanou dávku. Dávka se nemá zdvojnásobovat, aby se nahradila vynechaná dávka.
Zvláštní populace
Porucha funkce jater
U pacientů s mírnou poruchou funkce jater (třídy A podle Childa a Pugha) není nutná žádná úprava dávky. U pacientů se středně závažnou poruchou funkce jater (třídy B podle Childa a Pugha) se doporučuje snížit dávku na dvě tablety ráno a jednu tabletu večer (celkovou denní dávku 600 mg lumakaftoru / 375 mg ivakaftoru; viz bod 5.2).
S použitím přípravku Orkambi u pacientů se závažnou poruchou funkce jater (třídy C podle Childa a Pugha) nejsou žádné zkušenosti, ale očekává se, že expozice bude vyšší než u pacientů se středně závažnou poruchou funkce jater. Proto se má přípravek Orkambi používat po zvážení rizik a přínosů léčby s opatrností, v maximální celkové denní dávce 400 mg lumakaftoru / 250 mg ivakaftoru, podávané j ako j edna tableta ráno a j edna tableta večer, případně méně (viz body 4.4, 4.8 a 5.2).
Souběžné použití inhibitorů CYP3A
Pokud se u pacientů, kteří užívají přípravek Orkambi, zahájí podávání inhibitorů CYP3A, není nutná žádná úprava dávky. Pokud se však zahajuje podávání přípravku Orkambi u pacientů, kteří užívají silné inhibitory CYP3A, má se na první týden léčby dávka snížit na jednu tabletu denně (celkovou denní dávku 200 mg lumakaftoru / 125 mg ivakaftoru), aby byl umožněn indukční účinek lumakaftoru v ustáleném stavu. Po uplynutí této doby se má pokračovat doporučenou denní dávkou.
Pokud se při užívání silných inhibitorů CYP3A podávání přípravku Orkambi přeruší na dobu delší než jeden týden a opět se zahájí, má se na první týden znovuzahájené léčby dávka přípravku Orkambi snížit na jednu tabletu denně. Po uplynutí této doby se má pokračovat doporučenou denní dávkou (viz bod 4.5).
Porucha funkce ledvin
U pacientů s mírnou až středně závažnou poruchou funkce ledvin není nutná žádná úprava dávky. Při používání přípravku Orkambi u pacientů se závažnou poruchou funkce ledvin (clearance kreatininu nižší nebo rovná 30 ml/min) nebo u pacientů v konečném stadiu renálního onemocnění se doporučuje postupovat s opatrností (viz body 4.4 a 5.2).
Pediatrická populace
Bezpečnost a účinnost přípravku Orkambi u dětí ve věku do 12 let nebyla dosud stanovena. Nejsou dostupné žádné údaje (viz bod 5.1).
Starší osoby
Bezpečnost a účinnost přípravku Orkambi u pacientů ve věku od 65 let nebyla hodnocena.
Způsob podání
K perorálnímu podání. Pacienty je nutné poučit, aby polykali tablety vcelku. Pacienti nesmí tablety žvýkat, lámat ani rozpouštět.
4.3 Kontraindikace
Hypersenzitivita na léčivé látky nebo na kteroukoli pomocnou látku uvedenou v bodě 6.1.
4.4 Zvláštní upozornění a opatření pro použití
Pacienti s CF, kteří jsou heterozygotními nosiči mutace F508del v genu CFTR Kombinace lumakaftor/ivakaftor není účinná u pacientů s CF, kteří jsou nosiči mutace F508del na jedné alele a současně na druhé alele nosiči mutace, o níž se předpokládá, že vede k nedostatečné tvorbě proteinu CFTR, nebo in vitro neodpovídá na ivakaftor (viz bod 5.1).
Pacienti s CF, kteří jsou nosiči mutace ovlivňující otevírání kanálu (třídy III) v genu CFTR Kombinace lumakaftor/ivakaftor nebyla testována u pacientů s CF, kteří jsou nosiči mutace ovlivňující otevírání kanálů (třídy III) v genu CFTR na jedné alele, s mutací nebo bez mutace F508del na druhé alele. Jelikož expozice ivakaftoru je při podávání v kombinaci s lumakaftorem velmi významně snížená, nemá se kombinace lumakaftor/ivakaftor u těchto pacientů používat.
Respirační příhody
Respirační příhody (např. hrudní diskomfort, dyspnoe a abnormální dýchání) byly častější v průběhu zahajování terapie lumakaftorem/ivakaftorem. Klinické zkušenosti u pacientů s předpokládanou hodnotou usilovné, sekundové vitální kapacity forced expiratory volume in 1 second, FEVi)
vyjádřené v procentech (percentpredictedFEVi, ppFEVi) < 40 jsou omezené a v průběhu zahajování terapie se doporučuje tyto pacienty dále sledovat (viz bod 4.8). Nejsou žádné zkušenosti se zahájením léčby lumakaftorem/ivakaftorem u pacientů s plicní exacerbací a proto to není vhodné.
Účinek na krevní tlak
U některých pacientů léčených lumakaftorem/ivakaftorem byl pozorován zvýšený krevní tlak.
V průběhu léčby se má krevní tlak pravidelně sledovat u všech pacientů (viz bod 4.8).
Pacienti s pokročilým onemocněním jater
U pacientů s CF mohou být přítomny abnormality funkce jater včetně pokročilého onemocnění jater.
U některých pacientů s CF a s pokročilým onemocněním jater užívajících lumakaftor/ivakaftor bylo hlášeno zhoršení funkce jater. Kombinace lumakaftor/ivakaftor se má používat u pacientů s pokročilým onemocněním jater s opatrností a pouze, pokud se předpokládá, že přínosy léčby převáží nad riziky. Pokud se u těchto pacientů používá lumakaftor/ivakaftor, mají se pacienti po zahájení léčby pečlivě sledovat a dávka se má snížit (viz body 4.2, 4.8 a 5.2).
Hepatobiliární příhody
U pacientů s CF užívajících lumakaftor/ivakaftor byly hlášeny zvýšené hladiny transamináz.
V některých případech byla tato zvýšení spojena se souběžným zvýšením hladiny celkového bilirubinu v séru.
Jelikož nelze vyloučit spojení s poškozením jater, doporučuje se před zahájením léčby lumakaftorem/ivakaftorem, každé 3 měsíce v průběhu prvního roku léčby a potom jednou za rok provádět funkční jaterní testy (ALT, AST a bilirubin). U pacientů se zvýšenými hladinami ALT, AST nebo bilirubinu v anamnéze je nutné zvážit častější sledování.
V případě významného zvýšení hladin ALT nebo AST se zvýšením nebo bez zvýšení hladiny bilirubinu (buď ALT nebo AST > 5* horní hranice normálních hodnot [upper limit of normal, ULN], nebo ALT nebo AST > 3* ULN s hladinou bilirubinu > 2* ULN) se má podávání lumakaftoru/ivakaftoru ukončit a mají se pečlivě provádět laboratorní vyšetření, dokud abnormality nevymizí. Po snížení zvýšených hladin transamináz je nutné zvážit přínosy a rizika dalšího podávání přípravku (viz body 4.2, 4.8 a 5.2).
Interakce s léčivými přípravky Substráty CYP3A
Lumakaftor je silným induktorem CYP3A. Podávání přípravku Orkambi může snížit systémovou expozici léčivých přípravků, které jsou substráty CYP3A, čímž se sníží jejich léčebný účinek. Souběžné podávání se senzitivními substráty CYP3A nebo substráty CYP3A s úzkým terapeutickým indexem se nedoporučuje (viz bod 4.5).
Kombinace lumakaftor/ivakaftor může výrazně snížit expozici hormonálních kontraceptiv, čímž se sníží jejich účinnost. Při souběžném podávání s přípravkem Orkambi se na hormonální kontraceptiva, zahrnující kontraceptiva perorální, injektabilní, transdermální a implantabilní, nemá spoléhat jako na účinnou metodu antikoncepce (viz bod 4.5).
Silné induktory CYP3A
Ivakaftor je substrátem CYP3A4 a CYP3A5. Použití lumakaftoru/ivakaftoru se silnými induktory CYP3A, např. rifampicinem, významně snižuje expozici ivakaftoru, což může snížit terapeutickou účinnost lumakaftoru/ivakaftoru. Souběžné podávání se silnými induktory CYP3A (např. rifampicinem, třezalkou tečkovanou [Hypericumperforatum]) se proto nedoporučuje (viz bod 4.5).
Porucha funkce ledvin
Při používání lumakaftoru/ivakaftoru u pacientů se závažnou poruchou funkce ledvin nebo u pacientů v konečném stadiu renálního onemocnění se doporučuje postupovat s opatrností (viz body 4.2 a 5.2).
Kataraktv
U pediatrických pacientů léčených ivakaftorem v monoterapii byly hlášeny případy získaného zákalu oční čočky bez vlivu na zrak. Přestože v některých případech byly přítomny další rizikové faktory (např. podávání kortikosteroidů a ozáření), nelze vyloučit možné riziko, které lze přisoudit ivakaftoru (viz bod 5.3). U pediatrických pacientů, u kterých se zahajuje léčba lumakaftorem/ivakaftorem, se doporučují vstupní a následná oftalmologická vyšetření.
Pacienti po transplantaci orgánů
Kombinace lumakaftor/ivakaftor nebyla testována u pacientů s CF, kteří podstoupili transplantaci orgánů. Proto se použití u pacientů po transplantaci nedoporučuje. Interakce s imunosupresivy viz bod 4.5.
4.5 Interakce s jinými léčivými přípravky a jiné formy interakce
Při podávání v monoterapii je lumakaftor silným induktorem CYP3A a ivakaftor slabým inhibitorem CYP3A. Jiné léčivé přípravky mohou mít při souběžném podávání potenciál ovlivnit lumakaftor/ivakaftor, a také kombinace lumakaftor/ivakaftor může ovlivnit jiné léčivé přípravky.
Potenciál jiných léčivých přípravků ovlivnit lumakaftor/ivakaftor
Inhibitory CYP3A
Souběžné podávání lumakaftoru/ivakaftoru s itrakonazolem, silným inhibitorem CYP3A, nemělo vliv na expozici lumakaftoru, ale 4,3násobně zvýšilo expozici ivakaftoru. Vzhledem k indukčnímu účinku lumakaftoru v ustáleném stavu na CYP3A se neočekává, že by čistá expozice ivakaftoru při souběžném podávání s inhibitorem CYP3A přesáhla jeho čistou expozici při podávání bez lumakaftoru, v dávce 150 mg každých 12 hodin, což je schválená dávka ivakaftoru v monoterapii.
Pokud se u pacientů, kteří užívají lumakaftor/ivakaftor, zahájí podávání inhibitorů CYP3A, není nutná žádná úprava dávky. Pokud se však podávání lumakaftoru/ivakaftoru zahajuje u pacientů užívajících silné inhibitory CYP3A, má se na první týden léčby dávka snížit na jednu tabletu denně (celkovou denní dávku 200 mg lumakaftoru / 125 mg ivakaftoru), aby byl umožněn indukční účinek lumakaftoru v ustáleném stavu. Po uplynutí této doby se má pokračovat doporučenou denní dávkou podle bodu 4.2. Pokud se podávání lumakaftoru/ivakaftoru přeruší na dobu delší než jeden týden a opět se zahájí, má se po dobu prvního týdne znovuzahájené léčby dávka snížit na jednu tabletu denně.
Při podávání se středně silnými nebo slabými inhibitory CYP3A není doporučena žádná úprava dávky.
Induktory CYP3A
Souběžné podávání lumakaftoru/ivakaftoru s rifampicinem, silným induktorem CYP3A, mělo minimální účinek na expozici lumakaftoru, ale snížilo expozici (plochu pod křivkou závislosti plazmatické koncentrace na čase, area under the plasma concentration-time curve, AUC) ivakaftoru o 57 %. Proto se souběžné podávání lumakaftoru/ivakaftoru a silných induktorů CYP3A nedoporučuje.
Při podávání se středně silnými nebo slabými induktory CYP3A není doporučena žádná úprava dávky.
Potenciál lumakaftoru/ivakaftoru ovlivnit jiné léčivé přípravky Substráty CYP3A
Lumakaftor je silným induktorem CYP3A. Ivakaftor je při podávání v monoterapii slabým inhibitorem CYP3A. Předpokládá se, že čistým účinkem terapie lumakaftorem/ivakaftorem bude silná indukce CYP3A. Proto může souběžné použití lumakaftoru/ivakaftoru a substrátů CYP3A snížit expozici těchto substrátů.
Substráty P-gp
In vitro studie naznačují, že lumakaftor má potenciál jak inhibovat, tak indukovat P-gp. Kromě toho bylo v klinické studii s ivakaftorem v monoterapii prokázáno, že ivakaftor je slabým inhibitorem P-gp. Proto může souběžné použití lumakaftoru/ivakaftoru a substrátů P-gp (např. digoxinu) změnit expozici těchto substrátů.
Substráty CYP2B6 a CYP2C
Interakce se substráty CYP2B6 a CYP2C nebyly v podmínkách in vivo studovány. In vitro studie naznačují, že lumakaftor má potenciál indukovat CYP2B6, CYP2C8, CYP2C9 a CYP2C19; inhibice CYP2C8 a CYP2C9 však byla pozorována také in vitro. Kromě toho in vitro studie naznačují, že ivakaftor může inhibovat CYP2C9. Proto může souběžné použití lumakaftoru/ivakaftoru změnit (tj. buď zvýšit nebo snížit) expozici substrátů CYP2C8 a CYP2C9, snížit expozici substrátů CYP2C19, a značně snížit expozici substrátů CYP2B6.
Potenciál lumakaftoru/ivakaftoru interagovat s transportéry
Experimenty in vitro prokazují, že lumakaftor je substrátem proteinu rezistence karcinomu prsu (Breast Cancer Resistance Protein, BCRP). Souběžné podávání Orkambi s léčivými přípravky, které inhibují BCRP, může zvýšit plazmatickou koncentraci lumakaftoru. Lumakaftor inhibuje transportér organických aniontů (organic anion transporter, OAT) 1 a 3. Lumakaftor a ivakaftor jsou inhibitory BCRP. Souběžné podávání Orkambi s léčivými přípravky, které jsou substráty pro OAT1/3 a BCRP transport může zvýšit plazmatické koncentrace těchto léčivých přípravků. Lumakaftor a ivakaftor nejsou inhibitory OATP1B1, OATP1B3 a transportéru organických kationtů (organic cation transporter, OCT) 1 a 2. Ivakaftor není inhibitorem OAT1 a OAT3.
Potvrzené a jiné potenciálně významné lékové interakce
V tabulce 1 jsou uvedeny potvrzené nebo předpokládané účinky lumakaftoru/ivakaftoru na jiné léčivé přípravky nebo účinky jiných léčivých přípravků na lumakaftor/ivakaftor. Informace uvedené v tabulce jsou většinou získané ze studií in vitro. Doporučení uvedená ve sloupci „Klinická poznámka“ v tabulce 1 vycházejí ze studií lékových interakcí, klinické relevance nebo předpokládaných interakcí vzhledem k cestám eliminace. Na prvním místě jsou uvedeny lékové interakce, které mají nejvyšší klinickou relevanci.
|
Tabulka 1: Potvrzené a jiné potenciálně významné lékové interakce - doporučení týkající se dávky při použití lumakaftoru/ivakaftoru s jinými léčivými přípravky | ||
|
rp V r I l W V Třída souběžně podávaného léčiva: | ||
|
Název léčiva |
Účinek |
Klinická poznámka |
|
Souběžně podávané léčivé přípravky s nejvyšší klinickou relevancí | ||
|
Antialergika: | ||
|
montelukast |
~ LUM, IVA | |
|
i montelukast |
Není doporučena žádná úprava dávky | |
|
Kvůli indukčnímu |
montelukastu. Při souběžném podávání | |
|
účinku LUM na |
s lumakaftorem/ivakaftorem se má | |
|
CYP3A/2C8/2C9 |
provádět přiměřené klinické sledování podle uvážení. Kombinace lumakaftor/ivakaftor může snížit expozici montelukastu, což může snížit jeho účinnost. | |
|
fexofenadin | ||
|
~ LUM, IVA |
Pro dosažení požadovaných klinických | |
|
t nebo i fexofenadin |
účinků může být požadována úprava | |
|
Kvůli potenciální |
dávky fexofenadinu. Kombinace | |
|
indukci či inhibici P- |
lumakaftor/ivakaftor může měnit expozici | |
|
_EP_ |
fexofenadinu. | |
|
Tabulka 1: Potvrzené a jiné potenciálně významné lékové interakce - doporučení týkající se dávky při použití lumakaftoru/ivakaftoru s jinými léčivými přípravky | ||
|
rp V r I l W V Třída souběžně podávaného léčiva: Název léčiva |
Účinek |
Klinická poznámka |
|
Antibiotika: klarithromycin, telithromycin |
~ LUM t IVA Kvůli inhibičnímu účinku klarithromycinu, telithromycinu na CYP3A |
Při zahajování léčby klarithromycinem nebo telithromycinem u pacientů užívajících lumakaftor/ivakaftor není doporučena žádná úprava dávky lumakaftoru/ivakaftoru. |
|
4 klarithromycin, telithromycin Kvůli indukčnímu účinku LUM na CYP3A |
Při zahajování léčby lumakaftorem/ivakaftorem u pacientů užívajících klarithromycin nebo telithromycin se má dávka lumakaftoru/ivakaftoru po dobu prvního týdne snížit na jednu tabletu denně. | |
|
Má se zvážit alternativa k těmto antibiotikům, např. azithromycin. Kombinace lumakaftor/ivakaftor může snížit expozici klarithromycinu a telithromycinu, což může snížit jejich účinnost. | ||
|
erythromycin |
~ LUM t IVA Kvůli inhibičnímu účinku erythromycinu na CYP3A |
Při souběžném podávání s erythromycinem není doporučena žádná úprava dávky lumakaftoru/ivakaftoru. |
|
4 erythromycin Kvůli indukčnímu účinku LUM na CYP3A |
Má se zvážit alternativa k erythromycinu, např. azithromycin. Kombinace lumakaftor/ivakaftor může snížit expozici erythromycinu, což může snížit jeho účinnost. | |
|
Antiepileptika: karbamazepin, fenobarbital, fenytoin |
~ LUM 4 IVA Kvůli indukčnímu účinku těchto antiepileptik na CYP3A | |
|
4 karbamazepin, fenobarbital, fenytoin Kvůli indukčnímu účinku LUM na CYP3A |
Souběžné použití lumakaftoru/ivakaftoru s těmito antiepileptiky se nedoporučuje. Expozice ivakaftoru a antikonvulziva může být významně snížena, což může snížit účinnost obou léčivých látek. | |
|
Tabulka 1: Potvrzené a jiné potenciálně významné lékové interakce - doporučení týkající se dávky při použití lumakaftoru/ivakaftoru s jinými léčivými přípravky | ||
|
rp V r I l W V Třída souběžně podávaného léčiva: Název léčiva |
Účinek |
Klinická poznámka |
|
Antimykotika: itrakonazol*, ketokonazol, posakonazol, vorikonazol |
~ LUM t IVA Kvůli inhibičnímu účinku těchto antimykotik na CYP3A |
Při zahajování léčby těmito antimykotiky u pacientů užívajících lumakaftor/ivakaftor není doporučena žádná úprava dávky lumakaftoru/ivakaftoru. |
|
i itrakonazol, ketokonazol, vorikonazol Kvůli indukčnímu účinku LUM na CYP3A |
Při zahajování léčby lumakaftorem/ivakaftorem u pacientů užívajících tato antimykotika se má dávka lumakaftoru/ivakaftoru po dobu prvního týdne snížit na jednu tabletu denně. | |
|
i posakonazol Kvůli indukčnímu účinku LUM na UGT |
Souběžné použití lumakaftoru/ivakaftoru s těmito antimykotiky se nedoporučuje. Pokud je nezbytné tyto léky podávat, je nutné pacienty pečlivě sledovat kvůli průlomovým mykotickým infekcím. Kombinace lumakaftor/ivakaftor může snížit expozici těchto antimykotik, což může snížit jejich účinnost. | |
|
flukonazol |
~ LUM t IVA Kvůli inhibičnímu účinku flukonazolu na CYP3A |
Při souběžném podávání s flukonazolem není doporučena žádná úprava dávky lumakaftoru/ivakaftoru. |
|
i flukonazol Kvůli indukčnímu účinku LUM; flukonazol se vylučuje převážně renální exkrecí v nezměněné formě; v přítomnosti silných induktorů však bylo pozorováno mírné snížení expozice flukonazolu |
Může být nutná vyšší dávka flukonazolu, aby bylo dosaženo požadovaného klinického účinku. Kombinace lumakaftor/ivakaftor může snížit expozici flukonazolu, což může snížit jeho účinnost. | |
|
Tabulka 1: Potvrzené a jiné potenciálně významné lékové interakce - doporučení týkající se dávky při použití lumakaftoru/ivakaftoru s jinými léčivými přípravky | ||
|
rp V r I l W V Třída souběžně podávaného léčiva: Název léčiva |
Účinek |
Klinická poznámka |
|
Protizánětlivá léčiva: ibuprofen |
~ LUM, IVA | |
|
4 ibuprofen Kvůli indukčnímu účinku LUM na CYP3A/2C8/2C9 |
Může být nutná vyšší dávka ibuprofenu, aby bylo dosaženo požadovaného klinického účinku. Kombinace lumakaftor/ivakaftor může snížit expozici ibuprofenu, což může snížit jeho účinnost. | |
|
Antimykobakteriální léčiva: rifabutin, rifampicin*, rifapentin |
~ LUM 4 IVA Kvůli indukčnímu účinku antimykobakteriálních léčiv na CYP3A | |
|
4 rifabutin Kvůli indukčnímu účinku LUM na CYP3A |
Souběžné použití lumakaftoru/ivakaftoru s těmito antimykobakteriálními léčivy se nedoporučuje. Expozice ivakaftoru bude snížená, což může snížit účinnost lumakaftoru/ivakaftoru. | |
|
Může být nutná vyšší dávka rifabutinu, aby bylo dosaženo požadovaného klinického účinku. Kombinace lumakaftor/ivakaftor může snížit expozici rifabutinu, což může snížit jeho účinnost. | ||
|
^ rifampicin, rifapentin | ||
|
Benzodiazepiny: midazolam, triazolam |
~ LUM, IVA | |
|
4 midazolam, triazolam Kvůli indukčnímu účinku LUM na CYP3A |
Souběžné použití lumakaftoru/ivakaftoru s těmito benzodiazepiny se nedoporučuje. Kombinace lumakaftor/ivakaftor sníží expozici midazolamu nebo triazolamu, což sníží jejich účinnost. | |
|
Hormonální kontraceptiva: ethinylestradiol, norethindron a jiné progestogeny |
4 ethinylestradiol, norethindron a jiné progestogeny Kvůli indukčnímu účinku LUM na CYP3A/UGT |
Na hormonální kontraceptiva, zahrnující kontraceptiva perorální, injikovatelná, transdermální a implantabilní, se při souběžném podávání s lumakaftorem/ivakaftorem nemá spoléhat jako na účinnou metodu antikoncepce. Kombinace lumakaftor/ivakaftor může snížit expozici hormonálních kontraceptiv, což může snížit jejich účinnost. |
|
Tabulka 1: Potvrzené a jiné potenciálně významné lékové interakce - doporučení týkající se dávky při použití lumakaftoru/ivakaftoru s jinými léčivými přípravky | ||
|
rp V r I l W V Třída souběžně podávaného léčiva: Název léčiva |
Účinek |
Klinická poznámka |
|
Imunosupresiva: cyklosporin, everolimus, sirolimus, takrolimus (používané po transplantaci orgánů) |
~ LUM, IVA 4 cyklosporin, everolimus, sirolimus, takrolimus Kvůli indukčnímu účinku LUM na CYP3A |
Souběžné použití lumakaftoru/ivakaftoru s těmito imunosupresivy se nedoporučuje. Kombinace lumakaftor/ivakaftor sníží expozici těchto imunosupresiv, což může snížit jejich účinnost. Použití lumakaftoru/ivakaftoru u pacientů po transplantaci orgánů nebylo testováno. |
|
Inhibitory protonové pumpy: esomeprazol, lansoprazol, omeprazol |
~ LUM, IVA 4 esomeprazol, lansoprazol, omeprazol Kvůli indukčnímu účinku LUM na CYP3A/2C19 |
Může být nutná vyšší dávka těchto inhibitorů protonové pumpy, aby bylo dosaženo požadovaného klinického účinku. Kombinace lumakaftor/ivakaftor může snížit expozici těchto inhibitorů protonové pumpy, což může snížit jejich účinnost. |
|
Byliny: třezalka tečkovaná (Hypericum perforatum) |
~ LUM 4 IVA Kvůli indukčnímu účinku třezalky tečkované na CYP3A |
Souběžné použití lumakaftoru/ivakaftoru s třezalkou tečkovanou se nedoporučuje. Expozice ivakaftoru bude snížená, což může snížit účinnost lumakaftoru/ivakaftoru. |
|
Jiné souběžně podávané léčivé přípravy s klinickou relevancí | ||
|
Antiarytmika: digoxin |
~ LUM, IVA | |
|
t nebo 4 digoxin Kvůli potenciální indukci nebo inhibici P-gp |
Je třeba sledovat sérové hladiny digoxinu a dávka se má titrovat, aby se dosáhlo požadovaného klinického účinku. Kombinace lumakaftor/ivakaftor může změnit expozici digoxinu. | |
|
Antikoagulancia: dabigatran |
~ LUM, IVA | |
|
t nebo 4 dabigatran Kvůli potenciální indukci nebo inhibici P-gp |
Při souběžném podávání s lumakaftorem/ivakaftorem má probíhat odpovídající klinické sledování. Pro dosažení požadovaných klinických účinků může být doporučena úprava dávky dabigatranu. Lumakaftor/ivakaftor může měnit expozici dabigatranu. | |
|
Tabulka 1: Potvrzené a jiné potenciálně významné lékové interakce - doporučení týkající se dávky při použití lumakaftoru/ivakaftoru s jinými léčivými přípravky | ||
|
rp V r I l W V Třída souběžně podávaného léčiva: | ||
|
Název léčiva |
Účinek |
Klinická poznámka |
|
warfarin | ||
|
~ LUM, IVA | ||
|
t nebo i warfarin |
Pokud je nutné souběžné podávání | |
|
Kvůli potenciálnímu |
warfarinu s lumakaftorem/ivakaftorem, má | |
|
indukčnímu nebo |
se sledovat mezinárodní normalizovaný | |
|
inhibičnímu účinku |
poměr (international normalised ratio, | |
|
LUM na CYP2C9 |
INR). Kombinace lumakaftor/ivakaftor může změnit expozici warfarinu. | |
|
Antidepresiva: | ||
|
citalopram, |
~ LUM, IVA | |
|
escitalopram, sertralin | ||
|
| citalopram, |
Může být nutná vyšší dávka těchto | |
|
escitalopram, sertralin |
antidepresiv, aby bylo dosaženo | |
|
Kvůli indukčnímu |
požadovaného klinického účinku. | |
|
účinku LUM na |
Kombinace lumakaftor/ivakaftor může | |
|
CYP3A/2C19 |
snížit expozici těchto antidepresiv, což může snížit jejich účinnost. | |
|
bupropion |
~ LUM, IVA | |
|
i bupropion |
Může být nutná vyšší dávka bupropionu, | |
|
Kvůli indukčnímu |
aby bylo dosaženo požadovaného | |
|
účinku LUM na |
klinického účinku. Kombinace | |
|
CYP2B6 |
lumakaftor/ivakaftor může snížit expozici bupropionu, což může snížit jeho účinnost. | |
|
Kortikosteroidy pro | ||
|
systémovou aplikaci: |
~ LUM, IVA | |
|
methylprednisolon, | ||
|
prednison |
| methylprednisolon, |
Může být nutná vyšší dávka těchto |
|
prednison |
kortikosteroidů pro systémovou aplikaci, | |
|
Kvůli indukčnímu |
aby bylo dosaženo požadovaného | |
|
účinku LUM na |
klinického účinku. Kombinace | |
|
CYP3A |
lumakaftor/ivakaftor může snížit expozici methylprednisolonu a prednisonu, což může snížit jejich účinnost. | |
|
Antagonisté | ||
|
H2-receptorů: |
~ LUM, IVA | |
|
ranitidin | ||
|
t nebo i ranitidinu |
Může být nutná úprava dávky ranitidinu, | |
|
Kvůli možné indukci |
aby bylo dosaženo požadovaného | |
|
nebo inhibici P-gp |
klinického účinku. Kombinace lumakaftor/ivakaftor může změnit expozici ranitidinu. | |
|
Tabulka 1: Potvrzené a jiné potenciálně významné lékové interakce - doporučení týkající se dávky při použití lumakaftoru/ivakaftoru s jinými léčivými přípravky | ||
|
rp V r I l W V Třída souběžně podávaného léčiva: Název léčiva |
Účinek |
Klinická poznámka |
|
Perorální antidiabetika: |
~ LUM, IVA | |
|
repaglinid |
i repaglinid Kvůli indukčnímu účinku LUM na |
Může být nutná vyšší dávka repaglinidu, aby bylo dosaženo požadovaného klinického účinku. Kombinace |
|
CYP3A/2C8 |
lumakaftor/ivakaftor může snížit expozici repaglinidu, což může snížit jeho účinnost. | |
|
Poznámka: \ = zvýšení, j |
= snížení, ^ = beze změny; LUM = lumakaftor; IVA = ivakaftor. | |
|
* Podle klinických studií lékových interakcí. Všechny ostatní uvedené lékové interakce jsou předpokládané. | ||
4.6 Fertilita, těhotenství a kojení
Údaje o podávání lumakaftoru/ivakaftoru těhotným ženám jsou omezené (méně než 300 ukončených těhotenství) nebo nejsou k dispozici. Studie vývojové a reprodukční toxicity s lumakaftorem a ivakaftorem na zvířatech nenaznačují přímé nebo nepřímé škodlivé účinky, přičemž účinky byly zaznamenány pouze s ivakaftorem při dávkách toxických pro matku (viz bod 5.3). Podávání lumakaftoru/ivakaftoru v těhotenství se z preventivních důvodů nedoporučuje, pokud klinický stav matky nevyžaduje léčbu lumakaftorem/ivakaftorem.
Kojení
Není známo, zda se lumakaftor a/nebo ivakaftor a metabolity vylučují do lidského mateřského mléka. Dostupné farmakokinetické údaje u zvířat prokázaly vylučování jak lumakaftoru, tak ivakaftoru do mléka laktujících samic potkana. Riziko pro kojené děti tedy nelze vyloučit. Na základě posouzení prospěšnosti kojení pro dítě a prospěšnosti léčby pro matku je nutno rozhodnout, zda přerušit kojení nebo ukončit/přerušit podávání lumakaftoru/ivakaftoru.
Fertilita
Lumakaftor neměl u samců a samic potkana žádný vliv na parametry fertility a reprodukční výkonnosti. Ivakaftor zhoršil u samců a samic potkana parametry fertility a reprodukční výkonnosti. Při dávkách < 100 mg/kg/den nebyly pozorovány žádné účinky na parametry samčí či samičí fertility a reprodukční výkonnosti (viz bod 5.3).
4.7 Účinky na schopnost řídit a obsluhovat stroje
Přípravek Orkambi nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
Ivakaftor, který je jednou z léčivých složek přípravku Orkambi, má malý vliv na schopnost řídit nebo obsluhovat stroje. Ivakaftor může způsobit závrať (viz bod 4.8).
Pacienti, u kterých se závrať při užívání přípravku Orkambi objeví, musí být poučeni, aby neřídili dopravní prostředky ani neobsluhovali stroje, dokud příznaky nevymizí.
4.8 Nežádoucí účinky
Shrnutí bezpečnostního profilu
Nejčastějšími nežádoucími účinky, které zaznamenali pacienti ve věku od 12 let užívající lumakaftor/ivakaftor v souhrnných, placebem kontrolovaných studiích fáze 3, byly dyspnoe (14,0 % oproti 7,8 % s placebem), průjem (11,0 % oproti 8,4 % s placebem) a nauzea (10,2 % oproti 7,6 % s placebem).
Mezi závažné nežádoucí účinky, které se objevily alespoň u 0,5 % pacientů, patřily hepatobiliární příhody, např. zvýšené hladiny transamináz, cholestatická hepatitida a jaterní encefalopatie.
Seznam nežádoucích účinků v tabulce
Nežádoucí účinky zaznamenané v klinických studiích s dobou léčby lumakaftorem/ivakaftorem 24 týdnů u pacientů ve věku od 12 let, kteří jsou homozygotními nosiči mutace F508del v genu CFTR, jsou uvedeny v tabulce 2 a jsou seřazeny podle tříd orgánových systémů, frekvence a nežádoucích účinků. Nežádoucí účinky pozorované u samotného ivakaftoru jsou také uvedeny v tabulce 2. Nežádoucí účinky jsou uvedeny podle klasifikace frekvencí podle databáze MedDRA: velmi časté (> 1/10), časté (> 1/100 až < 1/10), méně časté (> 1/1000 až < 1/100), vzácné (> 1/10 000 až <1/1000), velmi vzácné (< 1/10 000) a není známo (z dostupných údajů nelze určit).
Tabulka 2: Nežádoucí účinky u pacientů léčených lumakaftorem/ivakaftorem a u pacientů léčených pouze ivakaftorem
|
Třída orgánových systémů |
F rekvence |
Nežádoucí účinky |
|
Infekce a infestace |
velmi časté |
Nasofaryngitida* |
|
časté |
Infekce horních cest dýchacích, rinitida | |
|
Cévní poruchy |
méně časté |
Hypertenze |
|
Poruchy nervového systému |
velmi časté |
Bolest hlavy*, závrať* |
|
méně časté |
Hepatická encefalopatief | |
|
Poruchy ucha a labyrintu |
časté |
Bolest ucha*, ušní diskomfort*, tinitus*, hyperémie bubínku*, vestibulární porucha* |
|
méně časté |
Ušní kongesce* | |
|
Respirační, hrudní a mediastinální poruchy |
velmi časté |
Nosní kongesce*, dyspnoe |
|
časté |
Abnormální dýchání, orofaryngeální bolest, kongesce vedlejších nosních dutin*,rinorea, faryngální erytém | |
|
Gastrointestinální poruchy |
velmi časté | |
|
časté |
Flatulence, zvracení | |
|
Poruchy jater a žlučových cest |
časté |
Zvýšené hladiny transamináz |
|
méně časté |
Cholestatická hepatitida{ | |
|
Poruchy kůže a podkožní tkáně |
časté | |
|
Poruchy reprodukčního systému a prsu |
časté |
Nepravidelná menstruace, dysmenorea, metroragie, útvar v prsu* |
|
méně časté |
Menoragie, amenorea, polymenorea, zánět prsu*, gynekomastie*, porucha prsní bradavky*, bolest bradavky*, oligomenorea | |
|
Vyšetření |
velmi časté |
Bakterie ve sputu* |
|
méně časté |
Zvýšený krevní tlak |
*Nežádoucí účinky a jejich frekvence pozorované u pacientů s monoterapií ivakaftorem (složka Orkambi) v klinických studiích f 1 pacient ze 738 { 2 pacienti ze 738
Údaje o bezpečnosti od pacientů léčených lumakaftorem/ivakaftorem po dobu dalších 24 týdnů v dlouhodobé pokračovací studii bezpečnosti a účinnosti (studii 3) byly podobné jako údaje v placebem kontrolovaných studiích trvajících 24 týdnů (viz bod 5.1).
Popis vybraných nežádoucích účinků
Hepatobiliární příhody
V průběhu placebem kontrolovaných studií fáze 3 trvajících 24 týdnů byla incidence maximálních hladin transamináz (ALT nebo AST) > 8* ULN 0,8 % u pacientů užívajících lumakaftor/ivakaftor
a 0,5 % u pacientů užívajících placebo, > 5* ULN 2,0 %, resp. 1,9 % a > 3* ULN 5,2 %, resp. 5,1 %. Incidence nežádoucích účinků spojených s transaminázami byla 5,1 % u pacientů léčených lumakaftorem/ivakaftorem a 4,6 % u pacientů, kterým bylo podáváno placebo. U 7 pacientů, kteří byli léčeni lumakaftorem/ivakaftorem, se objevily závažné nežádoucí příhody spojené s játry se zvýšenými hladinami transamináz, přičemž u 3 z nich byla současně zvýšená hladina celkového bilirubinu. Po ukončení podávání lumakaftoru/ivakaftoru se hodnoty funkčních jaterních testů u všech pacientů vrátily k hodnotám na začátku studie nebo se výrazně zlepšily (viz bod 4.4).
Ze 7 pacientů s preexistující cirhózou a/nebo portální hypertenzí, kteří užívali lumakaftor/ivakaftor v placebem kontrolovaných studiích fáze 3 bylo zhoršení funkce jater se zvýšenými hladinami ALT, AST a bilirubinu a jaterní encefalopatií pozorováno u jednoho pacienta. Příhoda se objevila do 5 dnů od zahájení podávání kombinace lumakaftor/ivakaftor a vymizela po ukončení jejího podávání (viz bod 4.4).
Respirační příhody
V průběhu placebem kontrolovaných studií fáze 3 trvajících 24 týdnů byla incidence respiračních nežádoucích účinků (např. hrudního diskomfortu, dyspnoe a abnormálního dýchání) 26,3 % u pacientů léčených lumakaftorem/ivakaftorem v porovnání se 17,0 % pacientů, kteří užívali placebo. Incidence těchto příhod byla častější u pacientů s nižší hodnotou FEV1 před léčbou; 29,6 % pacientů s ppFEV1
< 70 a 37,7 % pacientů s ppFEV1 < 40 v porovnání s 21,0 % pacientů užívajících placebo s ppFEV1
< 70 a 21,4 % pacientů užívajících placebo s ppFEVi < 40. Přibližně tři čtvrtiny příhod začaly
v průběhu prvního týdne léčby a u většiny pacientů vymizely bez přerušení léčby. Většina příhod měla mírný až středně závažný charakter, nebyla vážná a nevedla k ukončení léčby (viz bod 4.4).
Abnormality menstruace
V průběhu placebem kontrolovaných studií fáze 3 trvajících 24 týdnů byla incidence příhod kombinovaných abnormalit menstruace (amenorey, dysmenorey, menoragie, nepravidelné menstruace, metroragie, oligomenorey a polymenorey) 9,9 % u pacientek léčených lumakaftorem/ivakaftorem
a 1,7 % u žen léčených placebem. Tyto menstruační příhody se objevovaly častěji v podskupině pacientek, které užívaly hormonální kontraceptiva (25,0 %) v porovnání s pacientkami, které hormonální kontraceptiva neužívaly (3,5 %; viz bod 4.5). Většina těchto účinků měla mírný nebo středně závažný charakter, nebyla vážná. U pacientek léčených lumakaftorem/ivakaftorem přibližně dvě třetiny těchto účinků vymizely a medián doby trvání byl 10 dnů.
Zvýšený krevní tlak
V průběhu placebem kontrolovaných studií fáze 3 trvajících 24 týdnů byly nežádoucí účinky spojené se zvýšeným krevním tlakem (např. hypertenze, zvýšený krevní tlak) hlášeny u 0,9 % (7/738) pacientů léčených lumakaftorem/ivakaftorem a u žádného z pacientů, kteří užívali placebo.
U pacientů, kteří byli léčeni lumakaftorem/ivakaftorem (průměrná hodnota systolického tlaku na začátku studie 114 mmHg a průměrná hodnota diastolického tlaku na začátku studie 69 mmHg), bylo maximální zvýšení průměrné hodnoty systolického, resp. diastolického krevního tlaku oproti počáteční hodnotě 3,1 mmHg, resp. 1,8 mmHg. U pacientů, kteří užívali placebo (průměrná hodnota systolického tlaku na začátku studie 114 mmHg a průměrná hodnota diastolického tlaku na začátku studie 69 mmHg) bylo maximální zvýšení průměrné hodnoty systolického, resp. diastolického krevního tlaku oproti počáteční hodnotě 0,9 mmHg, resp. 0,9 mmHg.
Podíl pacientů, u kterých byla alespoň dvakrát zaznamenána hodnota systolického krevního tlaku > 140 mmHg nebo hodnota diastolického krevního tlaku > 90 mmHg, byl 3,4 %, resp. 1,5 % u pacientů léčených lumakaftorem/ivakaftorem v porovnání s 1,6 % a 0,5 % u pacientů, kteří užívali placebo (viz bod 4.4).
Pediatrická populace
Údaje o bezpečnosti byly shromážděny od 194 pediatrických pacientů s CF ve věku od 12 do 17 let, kteří jsou homozygotními nosiči mutace F508del a kteří užívali lumakaftor/ivakaftor v placebem kontrolovaných studiích fáze 3. Bezpečnostní profil u těchto pediatrických pacientů odpovídá bezpečnostnímu profilu u dospělých pacientů.
Hlášení podezření na nežádoucí účinky
Hlášení podezření na nežádoucí účinky po registraci léčivého přípravku je důležité. Umožňuje to pokračovat ve sledování poměru přínosů a rizik léčivého přípravku. Žádáme zdravotnické pracovníky, aby hlásili podezření na nežádoucí účinky prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V.
4.9 Předávkování
Při předávkování přípravkem Orkambi není k dispozici žádné specifické antidotum. Léčba předávkování sestává z obecných podpůrných opatření zahrnujících sledování životních funkcí a klinického stavu pacienta.
Nežádoucí příhody, které se v období podávání vyšší než terapeutické dávky objevily s incidencí > 5 % v porovnání s obdobím podávání terapeutické dávky, byly bolest hlavy, generalizovaná vyrážka a zvýšené hladiny transamináz.
5. FARMAKOLOGICKÉ VLASTNOSTI
5.1 Farmakodynamické vlastnosti
Farmakoterapeutická skupina: Jiná léčiva respiračního systému, ATC kód: R07AX30 Mechanismus účinku
Protein CFTR je chloridový kanál přítomný na povrchu epitelových buněk mnoha orgánů. Mutace F508del ovlivňuje protein CFTR mnoha způsoby, zejména zapříčiněním defektu při tvorbě a přesunu proteinu v buňce, což snižuje množství proteinu CFTR na buněčném povrchu. Na povrch buňky se dostane pouze malé množství proteinu F508del-CFTR, které má nízkou pravděpodobnost otevření kanálu (defektní otevírání kanálu). Lumakaftor je korektor proteinu CFTR, který působí přímo na protein F508del-CFTR a zlepšuje jeho tvorbu a přesun v buňce, čímž dochází ke zvýšení množství funkčních proteinů CFTR na buněčném povrchu. Ivakaftor je potenciátor proteinu CFTR, který usnadňuje zvýšený transport chloridů potencováním pravděpodobnosti otevření kanálu proteinu CFTR na buněčném povrchu. Kombinovaným účinkem lumakaftoru a ivakaftoru je zvýšené množství a zvýšená funkčnost proteinu F508del-CFTR na buněčném povrchu, což vede ke zvýšenému transportu chloridových iontů. Přesné mechanismy, kterými lumakaftor zlepšuje tvorbu a přesun proteinu F508del-CFTR a ivakaftor potencuje protein F508del-CFTR, nejsou známy.
Farmakodynamické účinky
Účinky na množství chloridů v potu:
Ve dvojitě zaslepené, placebem kontrolované klinické studii fáze 2 u pacientů s CF ve věku od 18 let byly hodnoceny změny množství chloridů v potu v odpovědi na podávání samotného lumakaftoru nebo lumakaftoru v kombinaci s ivakaftorem. V této studii dokončilo léčbu samotným lumakaftorem v dávce 400 mg po 12 hodinách po dobu 28 dnů s následným přidáním ivakaftoru v dávce 250 mg po 12 hodinách po dobu dalších 28 dnů 10 pacientů (homozygotních nosičů mutace F508del-CFTR) a léčbu placebem dokončilo 25 pacientů (homozygotních nebo heterozygotních nosičů mutace F508del). Léčebný rozdíl mezi samotným lumakaftorem v dávce 400 mg po 12 hodinách a placebem, hodnocený jako průměrná změna množství chloridů v potu od začátku studie do 28. dne, byl při hodnotě -8,2 mmol/l (95% interval spolehlivosti [confidence interval, CI]: -14; -2) statisticky významný. Léčebný rozdíl mezi kombinací lumakaftoru v dávce 400 mg / ivakaftoru v dávce 250 mg po 12 hodinách a placebem, hodnocený jako průměrná změna množství chloridů v potu od začátku studie do 56. dne, byl při hodnotě -11 mmol/l (95% CI: -18; -4) statisticky významný.
Změny ve FEVf.
V této studii byly také hodnoceny změny v ppFEV1 v odpovědi na lumakaftor samotný nebo v kombinaci s ivakaftorem. Léčebný rozdíl mezi samotným lumakaftorem v dávce 400 mg po
12 hodinách a placebem, hodnocený jako průměrná absolutní změna ppFEVi, byl -4,6 procentních bodů (95% CI: -9,6, 0,4) od počátku do 28. dne, 4,2 procentních bodů (95% CI: -1,3, 9,7) od počátku do 56. dne a 7,7 procentních bodů (95% CI: 2,6, 12,8; statisticky významný) od 28. do 56. dne (po přidání ivakaftoru k monoterapii lumakaftorem).
Snížení tepové frekvence:
V průběhu placebem kontrolovaných studií fáze 3 trvajících 24 týdnů bylo maximální snížení tepové frekvence o 6 tepů za minutu oproti počáteční hodnotě pozorováno 1. a 15. den přibližně za 4 až
6 hodin po užití dávky. Po 15. dnu nebyla v těchto studiích tepová frekvence v období po užití dávky sledována. Od 4. týdne se u pacientů léčených lumakaftorem/ivakaftorem pohybovala změna průměrné tepové frekvence před podáním dávky v rozmezí od 1 do 2 tepů za minutu pod počáteční hodnotou. V průběhu léčby byl procentní podíl pacientů s hodnotami tepové frekvence < 50 tepů za minutu 11 % u pacientů léčených lumakaftorem/ivakaftorem v porovnání se 4,9 % u pacientů užívajících placebo.
Klinická, účinnost.
Studie u pacientů s CF, kteří jsou homozygotními nosiči mutace F508del v genu CFTR Účinnost kombinace lumakaftor/ivakaftor byla u pacientů s CF, kteří jsou homozygotními nosiči mutace F508del v genu CFTR, hodnocena ve dvou randomizovaných, dvojitě zaslepených, placebem kontrolovaných klinických studiích s 1108 klinicky stabilními pacienty s CF, z nichž 737 pacientů bylo randomizováno k podávání lumakaftoru/ivakaftoru. Pacienti v obou studiích byli randomizováni v poměru 1.1.1 k užívání lumakaftoru v dávce 600 mg jednou denně / ivakaftoru v dávce 250 mg po 12 hodinách, lumakaftoru v dávce 400 mg po 12 hodinách / ivakaftoru v dávce 250 mg po 12 hodinách nebo placeba. Pacienti užívali hodnocený lék s jídlem s obsahem tuku po dobu 24 týdnů. Kromě něj užívali svou předepsanou léčbu CF (např. bronchodilatancia, inhalační antibiotika, dornasu alfa a hypertonický roztok chloridu sodného). Pacienti z těchto studií byli vhodní k pokračování v prodloužené zaslepené studii.
Ve studii 1 bylo hodnoceno 549 pacientů s CF ve věku od 12 let (průměrném věku 25,1 roku) s hodnotou ppFEVi při skríningu mezi 40-90 (průměrná hodnota ppFEVi na začátku studie 60,7 [rozmezí. 31,1 až 94,0]). Ve studii 2 bylo hodnoceno 559 pacientů ve věku od 12 let (průměrném věku 25,0 let) s hodnotou ppFEV1 při skríningu mezi 40-90 (průměrná hodnota ppFEV1 na začátku studie 60,5 [rozmezí. 31,3 až 99,8]). Pacienti, kteří měli v anamnéze kolonizaci např. mikroorganismy Burkholderia cenocepacia, Burkholderia dolosa nebo Mycobacterium abscessus nebo u kterých byly zaznamenány 3 nebo více abnormálních výsledků funkčních jaterních testů (ALT, AST, AP, GGT > 3násobek ULN nebo hladina celkového bilirubinu > 2násobek ULN), byli vyloučeni.
Primárním cílovým parametrem účinnosti byla v obou studiích absolutní změna hodnoty ppFEV1 ve 24. týdnu oproti počáteční hodnotě. Další parametry účinnosti zahrnovaly relativní změnu hodnoty ppFEV1 oproti počáteční hodnotě, absolutní změnu indexu tělesné hmotnosti (body mass index, BMI) oproti počáteční hodnotě, absolutní změnu v respirační doméně revidovaného dotazníku pro cystickou fibrózu (cystic fibrosis questionnaire - revised, CFQ-R) oproti počáteční hodnotě, podíl pacientů, kteří ve 24. týdnu dosáhli relativní změny hodnoty ppFEV1 oproti výchozí hodnotě o > 5 %, a počet případů plicní exacerbace (zahrnující případy vyžadující hospitalizaci nebo intravenózní antibiotickou terapii) do 24. týdne.
V obou studiích vedla léčba lumakaftorem/ivakaftorem ke statisticky významnému zlepšení hodnoty ppFEV1 (tabulka 3). Průměrné zlepšení hodnoty ppFEV1 bylo na začátku (15. den) rychlé a bylo zachováno po celé léčebné období trvající 24 týdnů. Patnáctý den byl v souhrnných studiích 1 a 2 léčebný rozdíl průměrné absolutní změny (95% CI) hodnoty ppFEV1 oproti počáteční hodnotě mezi lumakaftorem v dávce 400 mg / ivakaftorem v dávce 250 mg po 12 hodinách a placebem
2,51 procentního bodu (P < 0,0001). Zlepšení hodnoty ppFEV1 byla pozorována bez ohledu na věk, závažnost onemocnění, pohlaví a geografickou oblast. Do studií fáze 3 s lumakaftorem/ivakaftorem
bylo zahrnuto 81 pacientů s počáteční hodnotou ppFEV1 < 40. Léčebný rozdíl v této podskupině byl srovnatelný s léčebným rozdílem u pacientů s hodnotou ppFEV1 > 40. Ve 24. týdnu byl v souhrnných studiích 1 a 2 léčebný rozdíl průměrné absolutní změny (95% CI) hodnoty ppFEV1 oproti počáteční hodnotě mezi lumakaftorem v dávce 400 mg / ivakaftorem v dávce 250 mg po 12 hodinách a placebem 3,39 procentního bodu (P = 0,0382) u pacientů s hodnotou ppFEV1 < 40 a 2,47 procentního bodu (P < 0,0001) u pacientů s hodnotou ppFEVi > 40.
Tabulka 3: Shrnutí primárních a důležitých sekundárních výsledků ve studii ^ 1 a studii 2*
|
Studie 1 |
Studie 2 |
Souhrnný výsledek (studie 1 a studie 2) | |||||
|
Placebo (n = 184) |
LUM 400 mg po12 hod / IVA 250 mg po12 hod (n = 182) |
Placebo (n = 187) |
LUM 400 mg po 12 hod / IVA 250 mg po12 hod (n = 187) |
Placebo (n = 371) |
LUM 400 mg po 12 hod / IVA 250 mg po 12 hod (n = 369) | ||
|
Absolutní změna hodnoty ppFEV1 ve 24. týdnu (procentní body) |
Léčebný rozdíl |
- |
2,41 (P = 0,0003) f |
- |
2,65 (P = 0,0011) f |
- |
2,55 (P < 0,0001) |
|
Změna v rámci skupiny |
-0,73 (P = 0,216 8) |
1,68 (P = 0,0051) |
-0,02 (P = 0,973 0) |
2,63 (P < 0,0001) |
-0,39 (P < 0,349 4) |
2,16 (P < 0,0001) | |
|
Relativní změna hodnoty ppFEV 1 ve 24. týdnu (%) |
Léčebný rozdíl |
- |
4,15 (P = 0,0028)f |
- |
4,69 (P = 0,0009)f |
- |
4,4 (P < 0,0001) |
|
Změna v rámci skupiny |
-0,85 (P = 0,393 4) |
3,3 (P = 0,0011) |
0,16 (P = 0,879 3) |
4,85 (P < 0,0001) |
-0,34 (P = 0,637 5) |
4,1 (P < 0,0001) | |
|
Absolutní změna BMI ve 24. týdnu (kg/m2) |
Léčebný rozdíl |
- |
0,13 (P = 0,1938) |
- |
0,36 (P < 0,0001)f |
- |
0,24 (P = 0,0004) |
|
Změna v rámci skupiny |
0,19 (P = 0,006 5) |
0,32 (P < 0,0001) |
0,07 (P = 0,289 2) |
0,43 (P < 0,0001) |
0,13 (P = 0,006 6) |
0,37 (P < 0,0001) | |
|
Absolutní změna skóre v respirační doméně CFQ-R ve 24. týdnu (body) |
Léčebný rozdíl |
- |
1,5 (P = 0,3569) |
- |
2,9 (P = 0,0736) |
- |
2,2 (P = 0,0512) |
|
Změna v rámci skupiny |
1,1 (P = 0,342 3) |
2,6 (P = 0,0295) |
2,8 (P = 0,015 2) |
5,7 (P < 0,0001) |
1,9 (P = 0,021 3) |
4,1 (P < 0,0001) | |
|
Podíl pacientů s relativní změnou hodnoty ppFEV1 ve 24. týdnu o > 5 % |
% |
25 % |
32 % |
26 % |
41 % |
26 % |
37 % |
|
Poměr šancí |
- |
1,43 (P = 0,1208) |
- |
1,90 (P = 0,0032) |
- |
1,66 (P = 0,0013) | |
|
Počet případů plicní exacerbace do 24. týdne |
Počet příhod (poměrný výskyt za 48 týdnů) |
112 (1,07) |
73 (0,71) |
139 (1,18) |
79 (0,67) |
251 (1,14) |
152 (0,70) |
|
Relativní riziko |
- |
0,66 (P = 0,0169) |
- |
0,57 (P = 0,0002) |
- |
0,61 (P < 0,0001) | |
*V každé studii byl proveden hierarchický postup testování primárních a sekundárních cílových parametrů v každé aktivní léčebné skupině oproti placebu; pro statistickou významnost byla v každém kroku vyžadována hodnota P < 0,0250; tato hladina významnosti musela být dosažena také ve všech předcházejících testech.
^Indikuje statistickou významnost potvrzenou v hierarchickém postupu testování.
Ve 24. týdnu byl podíl pacientů, u kterých nedošlo k plicní exacerbaci, významně vyšší ve skupině léčené lumakaftorem/ivakaftorem v porovnání s placebem. V souhrnné analýze bylo relativní riziko exacerbace do 24. týdne u subjektů léčených lumakaftorem/ivakaftorem (lumakaftorem v dávce 400 mg / ivakaftorem v dávce 250 mg po 12 hodinách; n = 369) 0,61 (P < 0,0001), což představuje snížení o 39 % oproti placebu. Poměrný výskyt příhod za rok, anualizovaný na 48 týdnů, byl 0,70 ve skupině s lumakaftorem/ivakaftorem a 1,14 ve skupině s placebem. Léčba lumakaftorem/ivakaftorem v porovnání s placebem významně snížila riziko exacerbace vyžadující hospitalizaci o 61 % (relativní riziko = 0,39; P < 0,0001; poměrný výskyt příhod za 48 týdnů 0,17 ve skupině s lumakaftorem/ivakaftorem a 0,45 ve skupině s placebem) a snížila počet případů exacerbace vyžadujících léčbu intravenózními antibiotiky o 56 % (relativní riziko = 0,44; P < 0,0001; poměrný výskyt příhod za 48 týdnů 0,25 ve skupině s lumakaftorem/ivakaftorem a 0,58 ve skupině s placebem). Tyto výsledky nejsou považovány za statisticky významné v rámci hierarchie testování v jednotlivých studiích.
Dlouhodobá pokračovací studie bezpečnosti a účinnosti
Studie 3 je multicentrická, pokračovací studie fáze 3 s paralelními skupinami u pacientů s CF, do které byli zařazeni pacienti ze studie 1 a studie 2. Z 1108 pacientů, kteří dostávali jakoukoli léčbu ve studii 1 nebo studii 2, bylo do studie 3 zařazeno a aktivně léčeno 1029 (93 %) pacientů. Tato studie trvající 96 týdnů je zaměřena na hodnocení bezpečnosti a účinnosti dlouhodobé léčby lumakaftorem/ivakaftorem a stále pokračuje.
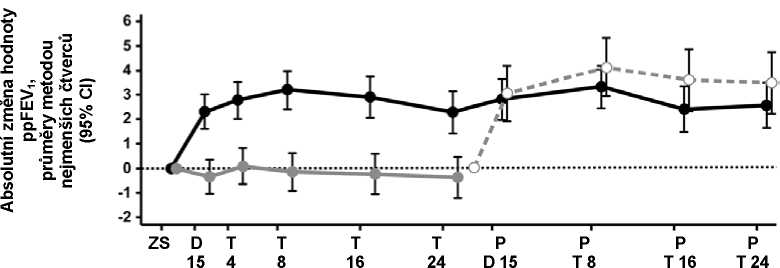
Jakmile všichni pacienti, kteří byli ve studii 1 nebo ve studii 2 léčeni lumakaftorem/ivakaftorem (lumakaftorem v dávce 400 mg / ivakaftorem v dávce 250 mg po 12 hodinách), absolvovali ve studii 3 návštěvu ve 24. týdnu (až 48 týdnů léčby), byla provedena ad hoc analýza účinnosti. Zlepšené hodnoty ppFEV1, pozorované u pacientů léčených lumakaftorem/ivakaftorem ve studii 1 nebo ve studii 2, zůstaly ve studii 3 zachovány (obrázek 1). Ve 24. týdnu studie 3 představovala zlepšení hodnot ppFEV1 oproti počáteční hodnotě ve studii 1 nebo ve studii 2 absolutní změnu o 2,6 procentního bodu a relativní změnu o 4,7 %. U pacientů léčených ve studii 1 nebo ve studii 2 lumakaftorem/ivakaftorem po dobu 24 týdnů vykazovali i po dalších 24 týdnech zlepšení hodnoty BMI. Průměrná absolutní změna BMI ve 24. týdnu studie 3 v porovnání s počátečními hodnotami ve studii 1 nebo ve studii 2 byla 0,56 kg/m2 (P < 0,0001).
Kromě toho 15. den léčby lumakaftorem/ivakaftorem (lumakaftorem v dávce 400 mg / ivakaftorem v dávce 250 mg po 12 hodinách) ve studii 3 byly u pacientů léčených placebem po dobu 24 týdnů ve studii 1 nebo ve studii 2 zaznamenány absolutní změny hodnoty ppFEV1 o 3,0 procentní body a relativní změna o 4,8 % v porovnání se začátkem studie 3, které jsou podobné zlepšením pozorovaným v aktivně léčených skupinách ve studii 1 a ve studii 2. Průměrná absolutní změna BMI ve 24. týdnu studie 3 v porovnání s počátečními hodnotami ve studii1 nebo ve studii 2 u pacientů, kteří byli původně léčeni placebem a potom lumakaftorem/ivakaftorem (lumakaftorem v dávce 400 mg / ivakaftorem v dávce 250 mg po 12 hodinách) byla 0,34 kg/m2 (P < 0,0001).
Obrázek 1. Absolutní změna předpokládané hodnoty FEV1 vyjádřené v procentech oproti počáteční hodnotě při každé návštěvě ve studii 3
Návštěva
Placebo
LUM 400 mg po 12 hod / IVA 250 mg po 12 hod -O - Placebo/LUM 400 mg po 1 2 hod / IVA 250 mg po 12 hod
Dlouhodobé údaje také prokazují, že časnější zahájení léčby lumakaftorem/ivakaftorem (lumakaftorem v dávce 400 mg / ivakaftorem v dávce 250 mg po 12 hodinách) snižuje poměrný výskyt plicní exacerbace. U pacientů léčených lumakaftorem/ivakaftorem po dobu až 48 týdnů byl poměrný výskyt příhod plicní exacerbace za rok (0,64; 95% CI: 0,55; 0,76) nižší než u pacientů, kteří byli 24 týdnů léčeni placebem ve studii 1 nebo ve studii 2 a potom po dobu až 24 týdnů lumakaftorem/ivakaftorem ve studii 3 (0,96; 95% CI: 0,79; 1,17).
Studie u pacientů s CF, kteří jsou heterozygotními nosiči mutace F508del v genu CFTR Studie 4 byla multicentrická, dvojitě zaslepená, randomizovaná, placebem kontrolovaná studie fáze 2 se 125 pacienty s CF ve věku od 18 let, u kterých byly zjištěny hodnoty ppFEVj 40-90 včetně a kteří jsou nosiči mutace F508del na jedné alele a současně jsou na druhé alele nosiči mutace, o níž se předpokládá, že vede k nedostatečné tvorbě proteinu CFTR, nebo in vitro neodpovídá na ivakaftor.
Pacienti byli léčeni buď lumakaftorem/ivakaftorem (n = 62), nebo placebem (n = 63) souběžně s jejich předepsanou léčbou CF. Primárním cílovým parametrem bylo zlepšení funkce plic, stanovené jako průměrná absolutní změna hodnoty ppFEV1 56. den v porovnání s počáteční hodnotou. Léčba lumakaftorem/ivakaftorem nevedla u pacientů s CF, kteří jsou heterozygotními nosiči mutace F508del v genu CFTR, k významnému zlepšení hodnoty ppFEV 1 v porovnání s placebem (léčebný rozdíl 0,60 [P = 0,5978]) ani k významnému zlepšení BMI nebo tělesné hmotnosti (viz bod 4.4).
Pediatrická populace
Evropská agentura pro léčivé přípravky udělila odklad povinnosti předložit výsledky studií s přípravkem Orkambi u jedné nebo více podskupin pediatrické populace s cystickou fibrózou. Informace o použití u dětí viz bod 4.2.
5.2 Farmakokinetické vlastnosti
Expozice (AUC) lumakaftoru je přibližně 2krát větší u zdravých dospělých dobrovolníků v porovnání s expozicí u pacientů s CF. Expozice ivakaftoru je u zdravých dospělých dobrovolníků a pacientů s CF podobná. Po podávání dvakrát denně bylo ustálených plazmatických koncentrací lumakaftoru a ivakaftoru u zdravých subjektů obvykle dosaženo přibližně po 7 dnech léčby, s poměrem akumulace přibližně 1,9 ve prospěch lumakaftoru. Expozice ivakaftoru v ustáleném stavu je nižší než expozice v ustáleném stavu 1. den kvůli indukčnímu účinku lumakaftoru na CYP3A (viz bod 4.5).
Po perorálním podávání lumakaftoru v dávce 400 mg po 12 hodinách / ivakaftoru v dávce 250 mg po 12 hodinách po jídle byly průměry v ustáleném stavu (± směrodatná odchylka [standard deviation, SD]) pro AUC0-12h 198 (64,8) pg-hod/ml a pro Cmax 25,0 (7,96) pg/ml u lumakaftoru a pro AUC0-12h 3,66 (2,25) pg-hod/ml a pro Cmax 0,602 (0,304) pg/ml u ivakaftoru. Po perorálním podávání samotného ivakaftoru v dávce 150 mg po 12 hodinách po jídle byly průměry v ustáleném stavu (±SD) pro AUC0-J2h 9,08 (3,20) pg-hod/ml a pro Cmax 1,12 (0,319) pg/ml.
Absorpce
Po opakovaných perorálních dávkách lumakaftoru se expozice lumakaftoru obvykle zvýšila úměrně dávce v rozmezí od 50 mg do 1000 mg každých 24 hodin. Expozice lumakaftoru se zvýšila přibližně 2,0násobně při podání s jídlem s obsahem tuku v porovnání s podáním nalačno. Medián (rozmezí) tmax lumakaftoru po jídle je přibližně 4,0 hodiny (2,0; 9,0).
Po opakovaném perorálním podání ivakaftoru v kombinaci s lumakaftorem se expozice ivakaftoru obvykle zvýšila při dávkách od 150 mg každých 12 hodin do 250 mg každých 12 hodin. Expozice ivakaftoru se při podání zdravým dobrovolníkům v kombinaci s lumakaftorem zvýšila přibližně 3násobně při podání s jídlem s obsahem tuku. Proto se má kombinace lumakaftor/ivakaftor podávat s jídlem s obsahem tuku. Medián (rozmezí) tmax ivakaftoru po jídle je přibližně 4,0 hodiny (2,0; 6,0).
Distribuce
Lumakaftor se přibližně z 99 % váže na plazmatické bílkoviny, zejména na albumin. Po perorálním podání dávky 400 mg každých 12 hodin pacientům s CF po jídle byly vypočtené typické zdánlivé distribuční objemy v centrálním a periferním kompartmentu (compartment volume, CV) 23,5 l (48,7 %), resp. 33,3 l (30,5 %).
Ivakaftor se přibližně z 99 % váže na plazmatické bílkoviny, zejména na alfa-1-kyselý glykoprotein a albumin. Po perorálním podání ivakaftoru v dávce 250 mg každých 12 hodin v kombinaci s lumakaftorem byly vypočtené typické zdánlivé distribuční objemy v centrálním a periferním kompartmentu (CV) 95,0 l (53,9 %), resp. 201 l (26,6 %).
In vitro studie naznačují, že lumakaftor je substrátem proteinu rezistence karcinomu prsu (BCRP).
Biotransformace
Lumakaftor u lidí nepodléhá rozsáhlému metabolismu, většina lumakaftoru se vylučuje v nezměněné formě stolicí. Údaje získané in vitro a in vivo naznačují, že lumakaftor se metabolizuje především oxidací a glukuronidací.
Ivakaftor u lidí podléhá rozsáhlému metabolismu. Údaje získané in vitro a in vivo naznačují, že ivakaftor se metabolizuje především prostřednictvím CYP3A. Dvěma hlavními metabolity ivakaftoru u lidí jsou metabolity M1 a M6. Účinnost metabolitu M1 odpovídá přibližně jedné šestině účinnosti ivakaftoru a tento metabolit je považován za farmakologicky účinný. Účinnost metabolitu M6 odpovídá přibližně jedné padesátině účinnosti ivakaftoru a tento metabolit není považován za farmakologicky aktivní.
Eliminace
Po perorálním podání lumakaftoru se většina lumakaftoru (51 %) vylučuje v nezměněné formě stolicí. Byla zaznamenána nepatrná exkrece lumakaftoru v nezměněné formě močí. Zdánlivý terminální poločas je přibližně 26 hodin. Vypočtená typická zdánlivá clearance (CL/F [CV]), lumakaftoru je u pacientů s CF 2,38 l/hod (29,4 %).
Po perorálním podání samotného ivakaftoru se většina ivakaftoru (87,8 %) eliminuje po metabolické konverzi stolicí. Byla zaznamenána nepatrná exkrece ivakaftoru v nezměněné formě močí. U zdravých subjektů je poločas ivakaftoru při podání s lumakaftorem přibližně 9 hodin. Vypočtená typická CL/F (CV) ivakaftoru při podání v kombinaci s lumakaftorem je u pacientů s CF 25,1 l/hod (40,5 %).
Porucha funkce jater
Po opakovaném podávání dávek lumakaftoru/ivakaftoru po dobu 10 dnů byly u subjektů se středně závažnou poruchou funkce jater (třídou B podle Childa a Pugha, skóre 7 až 9) zjištěny vyšší expozice (AUC0-J2h přibližně o 50 % a Cmax přibližně o 30 %) v porovnání se zdravými subjekty se stejnými demografickými údaji. Proto se má u pacientů se středně závažnou poruchou funkce jater (třídou B podle Childa a Pugha) dávka přípravku Orkambi snížit na dvě tablety ráno a jednu tabletu večer (celkovou denní dávku 600 mg lumakaftoru / 375 mg ivakaftoru). Vliv mírné poruchy funkce jater (třídy A podle Childa a Pugha, skóre 5 až 6) na farmakokinetiku lumakaftoru podávaného v kombinaci s ivakaftorem nebyl testován, ale lze očekávat, že zvýšení expozice bude menší než 50 %. U pacientů s mírnou poruchou funkce jater proto není nutná žádná úprava dávky.
U pacientů se závažnou poruchou funkce jater (třídy C podle Childa a Pugha, skóre 10 až 15) nebyly studie provedeny, ale lze očekávat, že expozice bude vyšší než u pacientů se středně závažnou poruchou funkce jater. U pacientů se závažnou poruchou funkce jater se má proto kombinace lumakaftor/ivakaftor užívat po zvážení rizik a přínosů léčby s opatrností, v maximální dávce jedna tableta ráno a jedna tableta večer (celkové denní dávce 400 mg lumakaftoru / 250 mg ivakaftoru), případně méně (viz body 4.2, 4.4 a 4.8).
Porucha funkce ledvin
Studie farmakokinetiky s lumakaftorem/ivakaftorem nebyly u pacientů s poruchou funkce ledvin provedeny. Ve studiích farmakokinetiky se samotným lumakaftorem byla u lidí zjištěna minimální eliminace lumakaftoru a jeho metabolitů močí (pouze 8,6 % celkové radioaktivity bylo zjištěno v moči, přičemž 0,18 % v nezměněné formě). Ve studii farmakokinetiky se samotným ivakaftorem byla u lidí zjištěna minimální eliminace ivakaftoru a jeho metabolitů močí (pouze 6,6 % celkové radioaktivity bylo zjištěno v moči). Populační farmakokinetická analýza clearance vs. clearance kreatininu neukazuje žádný trend pro subjekty s mírnou nebo středně závažnou poruchou funkce ledvin. Proto není při mírné nebo středně závažné poruše funkce ledvin doporučena žádná úprava dávky. Doporučuje se však postupovat s opatrností při podávání lumakaftoru/ivakaftoru pacientům se závažnou poruchou funkce ledvin (clearance kreatininu menší nebo rovná 30 ml/min) nebo u pacientů v konečném stadiu renálního onemocnění.
Starší lidé
Bezpečnost a účinnost lumakaftoru/ivakaftoru u pacientů ve věku od 65 let nebyla hodnocena.
Pohlaví
Vliv pohlaví na farmakokinetiku lumakaftoru byl hodnocen za použití populační farmakokinetické analýzy údajů z klinických studií s lumakaftorem podávaným v kombinaci s ivakaftorem. Výsledky naznačují, že mezi muži a ženami neexistuje ve farmakokinetických parametrech lumakaftoru nebo ivakaftoru klinicky relevantní rozdíl. Není nutná žádná úprava dávky přípravku Orkambi vzhledem k pohlaví.
5.3 Předklinické údaje vztahující se k bezpečnosti
Lumakaftor
Neklinické údaje získané na základě konvenčních farmakologických studií bezpečnosti, toxicity po opakovaném podávání, genotoxicity, hodnocení kancerogenního potenciálu, reprodukční a vývojové toxicity neodhalily žádné zvláštní riziko pro člověka. Zvláštní studie hodnotící fototoxický potenciál lumakaftoru nebyly provedeny; hodnocení dostupných neklinických a klinických údajů však nenaznačují žádnou fototoxickou zátěž.
Ivakaftor
Účinky ve studiích s opakovaným podáváním byly pozorovány pouze po expozicích dostatečně převyšujících (> 25násobně u myší, > 45násobně u potkanů a > 35násobně u psů) maximální expozici ivakaftoru, podávaného ve formě přípravku Orkambi, u člověka, což svědčí o malém významu při klinickém použití. Neklinické údaje získané na základě konvenčních studií genotoxicity a hodnocení kancerogenního potenciálu neodhalily žádné zvláštní riziko pro člověka.
Bezpečnostní farmakologie
Ivakaftor vykazoval na koncentraci závislý inhibiční účinek na koncové náboje lidského proteinu hERG (human ether-h-go-go relatedgene) při ICi5 5,5 pM, v porovnání s Cmax (1,5 pM) ivakaftoru při terapeutické dávce lumakaftoru/ivakaftoru. V telemetrické studii u psů při jednorázových dávkách až 60 mg/kg ani ve vyšetřeních EKG ze studií s opakovaným podáváním trvajících až 1 rok při dávkové hladině 60 mg/kg/den u psů (Cmax po 365 dnech = 36,2 až 47,6 pM) však nebylo pozorováno žádné prodloužení intervalu QT indukované ivakaftorem. Při jednorázových perorálních dávkách až 60 mg/kg u psů ivakaftor vyvolal na dávce závislé, avšak přechodné zvýšení parametrů krevního tlaku. V podrobné klinické studii hodnotící interval QT při podávání buď lumakaftoru v dávce 600 mg jednou denně / ivakaftoru v dávce 250 mg po 12 hodinách, nebo lumakaftoru v dávce 1000 mg jednou denně / ivakaftoru v dávce 450 mg po 12 hodinách nebyly pozorovány žádné významné změny intervalu QTc ani krevního tlaku, což prokazuje, že tato neklinická zjištění nelze přenášet do klinické praxe.
Těhotenství a fertilita
Ivakaftor nebyl teratogenní při perorálním podávání březím samicím potkana a králíka v průběhu organogenetického stadia fetálního vývoje v dávkách přibližně 10násobně (expozice ivakaftoru a metabolitů), resp. 46násobně převyšujících expozici ivakaftoru u člověka při terapeutické dávce lumakaftoru/ivakaftoru. Při dávkách toxických pro matku u potkanů způsobil ivakaftor snížení tělesné hmotnosti plodů; zvýšení incidence odchylek ve formě krčních žeber, hypoplastických žeber a volných žeber; a sternální abnormality zahrnující fúze. Významnost těchto zjištění pro člověka není známa.
Ivakaftor zhoršil u samců a samic potkana parametry fertility a reprodukční výkonnosti při dávkách 200 mg/kg/den (což představuje přibližně llnásobek, resp. 14násobek expozice dosažené při maximální doporučené dávce ivakaftoru jako složky přípravku Orkambi pro člověka, vycházející ze součtu hodnot AUC ivakaftoru a jeho metabolitů extrapolovaných z expozic dosažených 90. den při dávce 150 mg/kg/den ve studii toxicity po opakovaném podání trvající 6 měsíců, provedené u těchto druhů), při podávání samicím před zabřeznutím a v průběhu časné březosti. Při dávkách < 100 mg/kg/den (což představuje přibližně 8násobek expozice dosažené při maximální doporučené dávce ivakaftoru jako složky přípravku Orkambi pro člověka, vycházející ze součtu hodnot AUC
ivakaftoru a jeho metabolitů extrapolovaných z expozic dosažených 90. den při dávce 100 mg/kg/den ve studii toxicity po opakovaném podání trvající 6 měsíců, provedené u těchto druhů) nebyly pozorovány žádné účinky na parametry samčí a samičí fertility a reprodukční výkonnosti.
Perinatální a postnatální vývoj
Ivakaftor nezpůsobil vývojové vady u potomků březích samic potkana, kterým byly perorálně podávány dávky 100 mg/kg/den od březosti do porodu a odstavu. Dávky převyšující 100 mg/kg/den vyvolaly snížení parametrů přežití o 92 % a parametrů laktace o 98 %, stejně jako snížení tělesné hmotnosti mláďat.
Juvenilní zvířata
Nálezy katarakty byly pozorovány u juvenilních potkanů, kterým byly podávány dávky ivakaftoru 0,32násobně převyšující maximální doporučenou dávku pro člověka, vycházející ze systémové expozice ivakaftoru a jeho metabolitů při souběžném podávání s lumakaftorem ve formě přípravku Orkambi. Případy katarakty nebyly pozorovány u plodů samic potkana, které byly léčeny v průběhu stadia organogeneze fetálního vývoje, u mláďat potkana vystavených působení do určité míry sáním mléka před odstavením ani ve studiích toxicity po opakovaném podání ivakaftoru. Potenciální relevance těchto zjištění pro člověka není známa.
Lumakaftor a ivakaftor
Studie toxicity po opakovaném podání zahrnující souběžné podávání lumakaftoru a ivakaftoru neodhalily žádné zvláštní riziko pro člověka z hlediska potenciálu aditivní a/nebo synergické toxicity.
6. FARMACEUTICKÉ ÚDAJE
6.1 Seznam pomocných látek
Jádro tablety
Mikrokrystalická celulosa Sodná sůl kroskarmelosy Acetátosukcinát hypromelosy Povidon K30 Natrium-lauryl-sulfát Magnesium-stearát
Potahová vrstva Polyvinylalkohol Oxid titaničitý (E 171)
Makrogol 3350 Mastek
Karmín (E 120)
Hlinitý lak brilantní modře FCF (E 133) Hlinitý lak indigokarmínu (E 132)
Potiskový inkoust Šelak
Černý oxid železitý (E 172) Propylenglykol Hydroxid amonný
6.2 Inkompatibility
Neuplatňuje se.
6.3 Doba použitelnosti
2 roky
6.4 Zvláštní opatření pro uchovávání
Uchovávejte při teplotě do 30 °C.
6.5 Druh obalu a obsah balení
Blistr z PCTFE (polychlorotrifluoroethylenu) / PVC (polyvinylchloridu) s krytem z hliníkové fólie vyztužené papírem.
Přípravek Orkambi je dostupný v následujících velikostech balení:
Velikost balení: 112 tablet (4 balení po 28 tabletách).
Velikost balení: 56 tablet (2 balení po 28 tabletách).
Velikost balení: 28 tablet.
Na trhu nemusí být všechny velikosti balení.
6.6 Zvláštní opatření pro likvidaci přípravku
Žádné zvláštní požadavky.
7. DRŽITEL ROZHODNUTÍ O REGISTRACI
Vertex Pharmaceuticals (Europe) Limited
2 Kingdom Street
London
W2 6BD
Velká Británie
Tel: +44 (0) 1923 437672
8. REGISTRAČNÍ ČÍSLO(A)
EU/1/15/1059/001 EU/1/15/1059/002 EU/1/15/1059/003
9. DATUM PRVNÍ REGISTRACE / PRODLOUŽENÍ REGISTRACE
Datum první registrace: 19. listopadu 2015
10. DATUM REVIZE TEXTU
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
A. VÝROBCE ODPOVĚDNÝ ZA PROPOUŠTĚNÍ ŠARŽÍ
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
Název a adresa výrobce odpovědného za propouštění šarží
Almac Pharma Services Limited
Seagoe Industrial Estate
Craigavon
County Armagh
BT63 5UA
Velká Británie
B. PODMÍNKY NEBO OMEZENÍ VÝDEJE A POUŽITÍ
Výdej léčivého přípravku je vázán na lékařský předpis s omezením (viz příloha I: Souhrn údajů o přípravku, bod 4.2).
C. DALŠÍ PODMÍNKY A POŽADAVKY REGISTRACE • Pravidelně aktualizované zprávy o bezpečnosti
Požadavky pro předkládání pravidelně aktualizovaných zpráv o bezpečnosti pro tento léčivý přípravek jsou uvedeny v seznamu referenčních dat Unie (seznam EURD) stanoveném v čl. 107c odst. 7 směrnice 2001/83/ES a jakékoli následné změny jsou zveřejněny na evropském webovém portálu pro léčivé přípravky.
Držitel rozhodnutí o registraci předloží první pravidelně aktualizovanou zprávu o bezpečnosti pro tento léčivý přípravek do 6 měsíců od jeho registrace.
D. PODMÍNKY NEBO OMEZENÍ S OHLEDEM NA BEZPEČNÉ A ÚČINNÉ POUŽÍVÁNÍ LÉČIVÉHO PŘÍPRAVKU
• Plán řízení rizik (RMP)
Držitel rozhodnutí o registraci uskuteční požadované činnosti a intervence v oblasti farmakovigilance podrobně popsané ve schváleném RMP uvedeném v modulu 1.8.2 registrace a ve veškerých schválených následných aktualizacích RMP.
Aktualizovaný RMP je třeba předložit:
• na žádost Evropské agentury pro léčivé přípravky,
• při každé změně systému řízení rizik, zejména v důsledku obdržení nových informací, které mohou vést k významným změnám poměru přínosů a rizik, nebo z důvodu dosažení význačného milníku (v rámci farmakovigilance nebo minimalizace rizik).
• Povinnost uskutečnit poregistrační opatření:
Držitel rozhodnutí o registraci uskuteční v daném termínu níže uvedená opatření:
|
Popis |
Termín splnění |
|
Žadatel provede u pacientů s cystickou fibrózou pětiletou dlouhodobou pozorovací studii s lumakaftorem/ivakaftorem, která bude zahrnovat také mikrobiologické a klinické cílové parametry (např. exacerbace) podle schváleného protokolu. Žadatel předloží každoročně analýzy od prosince 2017 do prosince 2020 a konečnou zprávu klinické studie (CSR) do prosince 2021. |
Konečná zpráva klinické studie (CSR) v prosinci 2021 |
OZNAČENÍ NA OBALU A PŘÍBALOVÁ INFORMACE
A. OZNAČENÍ NA OBALU
Orkambi 200 mg/125 mg potahované tablety lumacaftorum/ivacaftorum
Jedna tableta obsahuje lumakaftorum 200 mg a ivakaftorum 125 mg.
112 potahovaných tablet (4 balení po 28 tabletách). 56 potahovaných tablet (2 balení po 28 tabletách). 28 potahovaných tablet.
Návod k použití
Ráno
Večer
Užívejte 2 tablety vcelku každých 12 hodin (ráno a večer) s jídlem s obsahem tuku (pokud Vám lékař nedal j iné pokyny).
Přípravek Orkambi můžete začít užívat kterýkoli den v týdnu.
Před použitím si přečtěte příbalovou informaci.
Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte při teplotě do 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Vertex Pharmaceuticals (Europe) Limited
2 Kingdom Street
London
W2 6BD
Velká Británie
Tel: +44 (0) 1923 437672
EU/1/15/1059/001 EU/1/15/1059/002 EU/1/15/1059/003
c.s.:
Orkambi
MINIMÁLNÍ ÚDAJE UVÁDĚNÉ NA BLISTRECH NEBO STRIPECH BLISTRY
1. NÁZEV LÉČIVÉHO PŘÍPRAVKU
Orkambi 200 mg/125 mg tablety lumacaftorum/ivacaftorum
2. NÁZEV DRŽITELE ROZHODNUTÍ O REGISTRACI
Vertex Pharmaceuticals (Europe) Limited
3. POUŽITELNOST
EXP
4. ČÍSLO ŠARŽE
č.š.:
5. JINÉ
Ráno
Večer
Orkambi 200 mg/125 mg potahované tablety lumacaftorum/ivacaftorum
Jedna tableta obsahuje lumakaftorum 200 mg a ivakaftorum 125 mg.
28 potahovaných tablet
5. ZPŮSOB A CESTA PODÁNÍ_
Návod k použití
Ráno
Večer
Užívejte 2 tablety vcelku každých 12 hodin (ráno a večer) s jídlem s obsahem tuku (pokud Vám lékař nedal j iné pokyny).
Přípravek Orkambi můžete začít užívat kterýkoli den v týdnu.
Před použitím si přečtěte příbalovou informaci.
Perorální podání
Uchovávejte mimo dohled a dosah dětí.
EXP
Uchovávejte při teplotě do 30 °C.
10. ZVLÁŠTNÍ OPATŘENÍ PRO LIKVIDACI NEPOUŽITÝCH LÉČIVÝCH PŘÍPRAVKŮ NEBO ODPADU Z NICH, POKUD JE TO VHODNÉ
Vertex Pharmaceuticals (Europe) Limited
2 Kingdom Street
London
W2 6BD
Velká Británie
Tel: +44 (0) 1923 437672
EU/1/15/1059/001-003
č.š.:
Orkambi
B. PŘÍBALOVÁ INFORMACE
Příbalová informace: informace pro pacienta
Orkambi 200 mg/125 mg potahované tablety
lumacaftorum/ivacaftorum
Tento přípravek podléhá dalšímu sledování. To umožní rychlé získání nových informací o bezpečnosti. Můžete přispět tím, že nahlásíte jakékoli nežádoucí účinky, které se u Vás vyskytnou. Jak hlásit nežádoucí účinky je popsáno v závěru bodu 4.
Přečtěte si pozorně celou příbalovou informaci dříve, než začnete tento přípravek užívat, protože obsahuje pro Vás důležité údaje.
- Ponechte si příbalovou informaci pro případ, že si ji budete potřebovat přečíst znovu.
- Máte-li jakékoli další otázky, zeptejte se svého lékaře nebo lékárníka.
- Tento přípravek byl předepsán výhradně Vám. Nedávejte jej žádné další osobě. Mohl by jí ublížit, a to i tehdy, má-li stejné známky onemocnění jako Vy.
- Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Viz bod 4.
Co naleznete v této příbalové informaci
1. Co je přípravek Orkambi a k čemu se používá
2. Čemu musíte věnovat pozornost, než začnete přípravek Orkambi užívat
3. Jak se přípravek Orkambi užívá
4. Možné nežádoucí účinky
5. Jak přípravek Orkambi uchovávat
6. Obsah balení a další informace
1. Co je přípravek Orkambi a k čemu se používá
Přípravek Orkambi se používá k dlouhodobé léčbě cystické fibrózy (CF) u pacientů ve věku od 12 let, kteří jsou nosiči specifické změny (nazývané mutace F508del), jenž ovlivňuje gen pro bílkovinu, která se označuje jako transmembránový regulátor vodivosti cystické fibrózy (cystic fibrosis transmembrane conductance regulátor, CFTR) a hraje významnou roli při regulaci toku hlenu v plicích. U lidí s touto mutací vzniká abnormální bílkovina CFTR. Buňky obsahují dvě kopie genu CFTR; přípravek Orkambi se používá u pacientů, kteří mají obě kopie postižené mutací F508del.
Přípravek Orkambi obsahuje dvě léčivé látky, lumakaftor a ivakaftor, které společně zlepšují funkci abnormální bílkoviny CFTR. Lumakaftor zvyšuje množství dostupné bílkoviny CFTR a ivakaftor pomáhá abnormální bílkovině fungovat normálnějším způsobem.
Během užívání přípravku Orkambi můžete zaznamenat, že se Vám lépe dýchá, že nejste tak často nemocní a/nebo, že snáze přiberete na váze.
2. Čemu musíte věnovat pozornost, než začnete přípravek Orkambi užívat Neužívejte přípravek Orkambi
- jestliže j ste alergický(á) na lumakaftor, ivakaftor nebo kteroukoli další složku tohoto přípravku (uvedenou v bodě 6).
Upozornění a opatření
Pokud Vám bylo sděleno, že máte onemocnění jater nebo ledvin, poraďte se se svým lékařem, protože může být nutné upravit dávku přípravku Orkambi.
U některých lidí užívajících přípravek Orkambi byly pozorovány abnormální výsledky jaterních testů (stanovené z krve). Okamžitě informujte svého lékaře, pokud se u Vás objeví kterýkoli z následujících příznaků, který může být známkou problémů s játry:
• Bolest nebo nepříjemný pocit v pravé horní části břicha
• Žloutnutí kůže nebo bělma očí
• Ztráta chuti k j ídlu
• Pocit na zvracení nebo zvracení
• Tmavě zbarvená moč
• Zmatenost
Lékař Vám bude v průběhu užívání přípravku Orkambi, zvlášť během prvního roku, provádět vyšetření krve, aby zkontroloval játra.
Respirační příhody, např. dušnost nebo tíseň na hrudi, byly u pacientů pozorovány na začátku užívání přípravku Orkambi. Pokud Vám plíce fungují špatně, může Vás lékař na začátku užívání přípravku Orkambi sledovat podrobněji.
U některých pacientů léčených přípravkem Orkambi bylo pozorováno zvýšení krevního tlaku.
V průběhu léčby přípravkem Orkambi může lékař sledovat Váš krevní tlak.
U některých dětí a adolescentů léčených ivakaftorem, složkou přípravku Orkambi, byla zaznamenána abnormalita oční čočky (zákal čočky neboli katarakta) bez jakéhokoli vlivu na zrak.
Před zahájením léčby přípravkem Orkambi a v jejím průběhu může Váš lékař provádět vyšetření očí.
Přípravek Orkambi nemají užívat jiní pacienti než ti, kteří mají dvě kopie F508del mutace v CFTR genu.
Přípravek Orkambi není doporučen pacientům, kteří podstoupili transplantaci orgánů.
Děti
Není známo, zda je přípravek Orkambi bezpečný a účinný při použití u dětí ve věku do 12 let. Proto se přípravek Orkambi nemá u dětí ve věku do 12 let používat.
Další léčivé přípravky a přípravek Orkambi
Informujte svého lékaře nebo lékárníka o všech lécích, které užíváte, které jste v nedávné době užíval(a) nebo které možná budete užívat.
Zejména informujte svého lékaře, pokud užíváte kterýkoli z následujících léků:
• Antibiotika (používaná k léčbě bakteriálních infekcí) např.: telithromycin, klarithromycin, rifampicin, rifabutin, rifapentin, erythromycin
• Antiepileptika (používaná k léčbě epileptických záchvatů) např.: fenobarbital, karbamazepin, fenytoin
• Benzodiazepiny (používané k léčbě úzkosti, nespavosti [insomnie], pohybového neklidu atd.) např. :
midazolam, triazolam
• Antimykotika (používaná k léčbě plísňových infekcí) např.: flukonazol, ketokonazol, itrakonazol, posakonazol, vorikonazol
• Imunosupresiva (používaná po transplantaci orgánů) např.: cyklosporin, everolimus, sirolimus, takrolimus
• Bylinné léky, např.:
třezalka tečkovaná (Hypericum perforatum)
• Antialergika (používaná k léčbě alergií a/nebo astmatu) např.: montelukast, fexofenadin
• Antidepresiva (používaná k léčbě deprese) např.: citalopram, escitalopram, sertralin, bupropion
• Protizánětlivé léky (používané k léčbě zánětu) např.: ibuprofen
• Antagonisté H2-receptorů (používané ke snížení hladiny žaludeční kyseliny) např.: ranitidin
• Srdeční glykosidy (používané k léčbě mírného nebo středně závažného městnavého srdečního selhání nebo abnormálního srdečního rytmu, který se označuje pojmem fibrilace síní) např.: digoxin
• Antikoagulancia (používaná k předcházení vzniku krevních sraženin nebo jejich zvětšování v krvi a krevních cévách) např.:
warfarin, dabigatran
• Antikoncepce (používaná k předcházení otěhotnění):
perorální (podávaná ústy), injikovatelná a implantabilní kontraceptiva, stejně jako antikoncepční náplasti; mohou obsahovat ethinylestradiol, norethindron a jiné progestogeny. Při společném používání s přípravkem Orkambi na ně nelze spoléhat jako na účinnou metodu kontroly početí
• Kortikosteroidy (používané k léčbě zánětu): methylprednisolon, prednison
• Inhibitory protonové pumpy (používané k léčbě refluxní choroby a vředů): omeprazol, esomeprazol, lansoprazol
• Perorální antidiabetika (používaná k léčbě cukrovky typu 2): repaglinid
Těhotenství a kojení
Pokud jste těhotná nebo kojíte, domníváte se, že můžete být těhotná, nebo plánujete otěhotnět, poraďte se se svým lékařem dříve, než začnete tento přípravek užívat. Pokud je to možné, je lepší se během těhotenství užívání přípravku Orkambi vyvarovat a Váš lékař Vám pomůže s rozhodnutím, co je pro Vás a Vaše dítě nejlepší.
Není známo, zda se lumakaftor nebo ivakaftor vylučují do lidského mateřského mléka. Pokud plánujete kojit, poraďte se se svým lékařem dříve, než začnete přípravek Orkambi užívat. Váš lékař rozhodne, zda přestanete kojit či zda ukončíte terapii lumakaftorem/ivakaftorem. Váš lékař bude brát v úvahu přínos kojení pro dítě a výhody terapie pro Vás.
Řízení dopravních prostředků a obsluha strojů
Přípravek Orkambi nemá žádný nebo má zanedbatelný vliv na schopnost řídit nebo obsluhovat stroje.
U pacientů užívajících ivakaftor, složku přípravku Orkambi, byla hlášena závrať, která může mít vliv na schopnost řídit nebo obsluhovat stroje. Pokud se u Vás závrať objeví, neřiďte dopravní prostředky ani neobsluhujte stroje, dokud tyto příznaky nevymizí.
Vždy užívejte tento přípravek přesně podle pokynů svého lékaře. Pokud si nejste jistý(á), poraďte se se svým lékařem.
Dávka
Doporučená dávka přípravku pro pacienty ve věku od 12 let je dvě tablety dvakrát denně (každých 12 hodin, celkem čtyři tablety [800 mg lumakaftoru / 500 mg ivakaftoru]) denně).
Jestliže máte středně závažné nebo závažné problémy s funkcí jater, může Vám lékař dávku přípravku Orkambi snížit, protože játra nebudou přípravek Orkambi vylučovat tak rychle jako u lidí s normální funkcí jater.
• Středně závažné problémy s játry: dávka může být snížena na dvě tablety ráno a jednu tabletu večer (celkem 600 mg lumakaftoru / 375 mg ivakaftoru denně).
• Závažné problémy s játry: dávka může být snížena na jednu tabletu (200 mg lumakaftoru /
125 mg ivakaftoru) každých 12 hodin.
Způsob podání
Přípravek Orkambi se užívá ústy.
Polykejte tablety vcelku. Tablety nežvýkejte, nelámejte ani nerozpouštějte.
Přímo před užitím přípravku Orkambi nebo ihned po jeho užití je třeba sníst jídlo nebo svačinu s obsahem tuku.
Užívání přípravku Orkambi s jídlem s obsahem tuku je důležité, aby bylo dosaženo správné hladiny léku v těle. Jídla a svačiny doporučené ve směrnicích pro CF nebo jídla doporučená ve standardních směrnicích pro výživu obsahují adekvátní množství tuku. Jídla nebo svačiny s obsahem tuku zahrnují například pokrmy připravené na másle nebo oleji nebo pokrmy obsahující vejce. Příklady jídel s obsahem tuku jsou:
• sýr, plnotučné mléko, mléčné výrobky z plnotučného mléka
• maso, tučné ryby
• avokádo, hummus, výrobky na bázi sóji (tofu)
• výživné tyčinky nebo nápoje
Jestliže jste užil(a) více přípravku Orkambi, než jste měl(a)
Poraďte se se svým lékařem nebo lékárníkem. Pokud je to možné, přineste s sebou svůj lék a tuto příbalovou informaci. Mohou se objevit nežádoucí účinky včetně těch, které jsou uvedeny níže v bodě 4.
Jestliže jste zapomněl(a) užít přípravek Orkambi
Pokud od doby, kdy jste dávku vynechal(a), uplynulo méně než 6 hodin, užijte vynechanou dávku ihned s jídlem obsahujícím tuk. V opačném případě počkejte do doby, kdy obvykle dávku užíváte. Nezdvojnásobujte následující dávku, abyste nahradil(a) vynechané tablety.
Jestliže jste přestal(a) užívat přípravek Orkambi
Užívejte lék stále podle pokynů svého lékaře, i když se cítíte lépe.
Máte-li jakékoli další otázky týkající se užívání tohoto přípravku, zeptejte se svého lékaře nebo lékárníka.
4. Možné nežádoucí účinky
Podobně jako všechny léky může mít i tento přípravek nežádoucí účinky, které se ale nemusí vyskytnout u každého.
Nežádoucí účinky hlášené při užívání přípravku Orkambi a samotného ivakaftoru (jedna z léčivých látek přípravku Orkambi) jsou uvedeny níže a mohou se objevit při užívání přípravku Orkambi.
Mezi závažné nežádoucí účinky přípravku Orkambi patří zvýšené hladiny jaterních enzymů v krvi, poškození jater a zmatenost spojená s nedostatečnou funkcí jater. Tyto závažné nežádoucí účinky jsou méně časté. Oznamte okamžitě svému lékaři, pokud máte významné příznaky jako bolest či nepříjemný pocit v horní oblasti žaludku (břicha), žloutnutí pokožky či bělma očí, ztrátu chuti k jídlu, pocit na zvracení či zvracení a tmavou moč.
Nežádoucí účinky přípravku Orkambi a ivakaftoru samotného (označeny hvězdičkou):
Velmi časté mohou postihnout více než 1 z 10 pacientů:
• bolest hlavy*
• bolest břicha (žaludku) *
• překrvení nosní sliznice*
• běžné nachlazení*
• dušnost
• změna typu bakterií v hlenu*
• závrať*
• pocit na zvracení
• průjem
Časté mohou postihnout až 1 z 10 pacientů:
• tíseň na hrudi
• překrvení sliznice vedlejších nosních dutin*
• infekce horních cest dýchacích
• bolest v krku
• ucpaný nos nebo rýma
• plynatost
• vyrážka
• zvracení
• začervenání v krku*
• nepravidelná menstruace nebo bolest při menstruaci
• bolest ucha, nepříjemný pocit v uchu*
• zvonění v uších*
• zarudnutí uvnitř ucha*
• hmatná bulka v prsu*
Méně časté mohou postihnout až 1 ze 100 pacientů:
• abnormální menstruace zahrnující nepřítomnost menstruace, prodloužené intervaly mezi cykly, zkrácené intervaly mezi cykly nebo silnější menstruační krvácení
• zvýšení krevního tlaku
• ušní kongesce*
• zánět prsu*
• zvětšení prsu*
• změny na prsních bradavkách nebo bolest prsních bradavek*
Hlášení nežádoucích účinků
Pokud se u Vás vyskytne kterýkoli z nežádoucích účinků, sdělte to svému lékaři nebo lékárníkovi. Stejně postupujte v případě jakýchkoli nežádoucích účinků, které nejsou uvedeny v této příbalové informaci. Nežádoucí účinky můžete hlásit také přímo prostřednictvím národního systému hlášení nežádoucích účinků uvedeného v Dodatku V. Nahlášením nežádoucích účinků můžete přispět k získání více informací o bezpečnosti tohoto přípravku.
Uchovávejte tento přípravek mimo dohled a dosah dětí.
Nepoužívejte tento přípravek po uplynutí doby použitelnosti uvedené na krabičce/blistru za EXP. Doba použitelnosti se vztahuje k poslednímu dni uvedeného měsíce.
Uchovávejte při teplotě do 30 °C.
Nevyhazujte žádné léčivé přípravky do odpadních vod nebo domácího odpadu. Zeptejte se svého lékárníka, jak naložit s přípravky, které již nepoužíváte. Tato opatření pomáhají chránit životní prostředí.
6. Obsah balení a další informace Co přípravek Orkambi obsahuje
Léčivými látkami jsou lumakaftorum a ivakaftorum. Jedna potahovaná tableta obsahuje lumacaftorum 200 mg a ivacaftorum 125 mg.
Dalšími složkami jsou:
• Jádro tablety: mikrokrystalická celulosa, sodná sůl kroskarmelosy, acetátosukcinát hypromelosy, povidon K30, natrium-lauryl-sulfát, magnesium-stearát
• Potahová vrstva: polyvinylalkohol, oxid titaničitý (E 171), makrogol 3350, mastek, karmín (E 120), hlinitý lak brilantní modře FCF (E 133), hlinitý lak indigokarmínu (E 132)
• Potiskový inkoust: šelak, černý oxid železitý (E 172), propylenglykol, hydroxid amonný Jak přípravek Orkambi vypadá a co obsahuje toto balení
Orkambi 200 mg/125 mg potahované tablety (tablety) jsou růžové tablety oválného tvaru (o rozměrech 14 x 8,4 x 6,8 mm) s černě vytištěným označením „2V125“ na jedné straně.
Přípravek Orkambi je dostupný v následujících velikostech balení:
Velikost balení: 112 tablet (4 balení po 28 tabletách).
Velikost balení: 56 tablet (2 balení po 28 tabletách).
Velikost balení: 28 tablet.
Na trhu nemusí být všechny velikosti balení.
Držitel rozhodnutí o registraci a výrobce
Vertex Pharmaceuticals (Europe) Limited
2 Kingdom Street
London
W2 6BD
Velká Británie
Tel: +44 (0) 1923 437672
Almac Pharma Services Limited
Seagoe Industrial Estate
Craigavon
County Armagh
BT63 5UA
Velká Británie
Tel: +44 (0) 28 3836 3363
Tato příbalová informace byla naposledy revidována Další zdroje informací
Podrobné informace o tomto léčivém přípravku jsou k dispozici na webových stránkách Evropské agentury pro léčivé přípravky http://www.ema.europa.eu.
41